

1. НАСЛЕДСТВЕНИ БОЛЕСТИ ПРИ ГОВЕДОТО
Наследствен дефицит в активността на ензима Алфа - манозидаза (Alpha-Mannosidosis). Алфа-манозидозата е наследствено автозомно-рецесивно наследяващо се заболяване с летален ефект. Въпреки, че Алфа-манозидозата (MA) е едно от най-отдавна известните на учените наследствени заболявания при породите Ангус, Мъри грей и Галоуей, то е малко познато сред фермерите, отглеждащи посочените породи говеда. Интересен е факта, че това заболяване не е открито при породите за мляко. По-голямата част от хомозиготните телета Ангус и Мъри грей умират скоро след раждане, а тези които все пак оцелеят показват тежко, прогресиращо неврологично заболяване, което се характеризира с тремор на главата, атаксия и агресия. Този факт показва, че заболяването се характеризира още с различна степен на експресия на мутантния алел. Фенотипно заболяването при породата Галоуей протича по-тежко и засегнатите телета се абортират или раждат мъртви. В Австралия заболяването се изучава усилено въпреки, че досега е събрана значително информация за разпространението му в популациите на посочените породи говеда. Изкореняването на МА е започнало още през 1980 година, когато тази диагноза е поставена на няколко животни. Разработването на ДНК тест за диагностика и откриване на хетерозиготни индивиди е дало точното средство за борба с МА при Ангуса в Австралия и САЩ. Изследвания са извършени от Angus Society of Australia върху 34203 говеда и резултатите показват, че 1863 животни (5,4%) са хетерозиготни и те са открити в 214 (51%) от изследваните стада.
Освен в Австралия заболяването е установено в САЩ и други държави. Предприети са и превантивни мерки чрез генотипизация на Червения Ангус в посочените държави.
ДНК теста се базира на PCR, който е разработен за две породо-специфични мутации отговорни за автозомно-рецесивно наследяващото се заболяване МА (Berg et al., 1997). Тестът включва отделна амплификация на два екзона на гена за лизозомна алфа-манозидаза последвана от рестрикционна дигестия на ампликоните. По този начин се показва, че една от мутациите (662G-->A заместване) е отговорна за МА при Глоуейското говедо. Друга мутация (961T-->C) заместване е уникално свързана с МА при породите Ангус, Мъри грей и Брангус в Австралия. Тази мутация е открита и при Червения Ангус внесен от Канада в Австралия под формата на ембриони. Допълнителни изследвания показват, че двете породо-специфични мутации може да увеличат честотата си на разпространение в Шотландия, а чрез износ на животни и замразена сперма в Америка, Нова Зеландия и Австралия.
Няма лечение за това заболяване. Единственият начин за борба е генотипизирането на одобрените за разплод бици, майките бикопроизводителки и елитната част от кравите в породата.
Литература.
1.Berg T., Healy P.J., Tollersrud O.K., Nilssen O. Molecular heterogeneity for bovine alpha-mannosidosis: PCR based assays for detection of breed-specific mutations. Res. Vet. Sci. 1997, 63(3):279-82.
2. Healy P.J., Babidge P.J., Embury D.H., Harrison M.A., Judson G.J., Mason R.W., Petterson D.S., Sinclair A.J. Control of alpha-mannosidosis in Angus cattle. Aust Vet J. 1983 May;60(5):135-7.
Наследствен дефицит в активността на ензима Бета - манозидaза (Beta-Mannosidоsis). Много наследствени дефицити на лизозомните ензими и свързаното с това натрупване на непреработени субстрати са добре характеризирани при човека и животните. Дефицитът на бета - манозидазата води до нарушение в преработката на специфичен полизахарид и това най-напред е установено при Нубийската коза и по-късно при човека и говедото (породата Салерс). При посочената порода говеда има заместваща мутация на G с A в позиция 2574 на комплементарната ДНК, която кодира последователности на незрелия стоп кодон близо до 3' края на протеин кодиращата част. За да се помогне на фермерите, отглеждащи говеда от породата Салерс е разработен PCR тест за откриване на носителите на тази мутация в гена, кодиращ ензима бета-манозидаза. Използването на този тест посочи наличието на два псевдо гена за бета-манозидаза Части от псевдогените са амплифицирани с алел-специфични праймери и след това секвенирани.
Бета-манозидозата се наблюдава при новородените телета от породата Салерс в Северна Америка и Нова Зеландия, но тази порода произхожда от Франция и е много вероятно това заболяване да съществува и там. Болните телета не растат нормално и показват клатещи движения на главата и тремор на други телесни части. Лицевите деформации включват прогнатизъм (издаване на горната или долната челюст), тесни цепки на клепачите и насочени назад уши. Установява се дисмиелинизация на централната нервна система, церебрални вентрикуларни разширения, намалено количество на плазмата, лимфоцитите, намалена активност на тъканната бета-манозидаза, отлагане на олигозахариди в тъканите и намалена концентрация на тироидния хормон в кръвния серум. Болните телета реагират на силен шум, имат запазен слухов потенциал и малки малки ушни и очни лезии. Тиромегалията и реномегалията са особено драматични при болните телета. Най-силно са засегнати проксималния бъбречен тубуларен епител (P2 сегмент), тироидния фоликуларен епител, много невронни субтипове и ретикулоендотелни клетки. На ултарструкторно ниво увеличените лизозоми са изпълнени с блестящи и тънки мембрани или мътно съдържание. Бялата материя в церебралната хемисфера е по-силно засегната от дисмиелинизацията отколкото клетките на главния и гръбначния мозък. Умереното и силно вентрикуларно разширение в главния мозък е по-скоро свързано със загуба на бяла материя отколкото с някакви обструкции. Аксоновите сфероидни изменения са добре изразени в оптичния нерв, бялото вещество в главния и малкия мозък, крайните участъци на триделния нерв и се дължат на отлагане на неразградени въглехидрати в изграждащите ги клетки. Основно това са олигозахариди като дизахарида ManP1-4GlcNAc и тризахарида Man,B1-4GlcNAc,B1-4GlcNAc. За да се улесни диагнозата на заболяването се използва тънкослойна хроматография на тъканни и уринарни олигозахариди. Диагнозата се поставя окончателно след определяне активността на бета-манозидазата в пламата или лимфоцитите. Понастоящем за борба с това наследствено заболяване се използва ДНК теста разработен от Leipprandt et al. (1999).
Литература
1. Margaret Z. Jones and Bruce Abbittt. Bovine beta-Mannosidosis.American journal of Pathology, Vol. 142, No. 3, March 1993
2. Leipprandt JR, Chen H, Horvath JE, Qiao XT, Jones MZ, Friderici KH. Identification of a bovine beta-mannosidosis mutation and detection of two beta-mannosidase pseudogenes. Mamm. Genome. 1999, 10(12):1137-41.
Артрогрипозис мултиплекс (Arthrogryposis Multiplex - АМ). AM при говедото е открит като генетичен дефект на 16.09.2008 г. Телетата са се раждали или мъртви или са умирали скоро след раждане. Гърбът и краката са били извити или изкривени в ставите и често са били фиксирани в тази позиция. Предните крайници са били свити и повдигнати или изпънати. Телетата са били малки по размер поради слабо развитата мускулатура. Понакога е имало разцепване на носната част или на горното небце. Ангус Асоцияцията в САЩ предлага този генетичен дефект при породата Ангус да се нарече Артрогрипозис мултиплекс (Arthrogryposis Multiplex - AM; aka; Curly Calf Syndrome). Изследователите потвърждават, че AM е летален генетичен дефект и че се наследява като автозомно-рецесивен признак. Известно е че при този начин за наследяване на признаците заболяването се проявява фенотипно само ако засегнатия индивид е хомозиготен по рецесивния алел. Хетерозиготните индивиди са само носители на мутантния алел и не боледуват, но са основен източник за разпространение на заболяването. Опасността от раждане на телета с този дефект пряко зависи от броя на хетерозиготните родители в популацията. Колкото по-голям е техния брой толкова по-голям е риска от раждане на болни телета. Статистически разпределението на индивидите в голяма популация е 25% хомозиготни по нормалния алел индивиди, 75% хетерозиготни и 25% хомозиготни по мутантния алел. При малки по обем популации това разпределение не може да се случи и всичко зависи от случайността при оплождане на яйцеклетката от сперматозоидите. Ако сперматозоид носител на мутантния алел оплоди яйцеклетка носител на същия алел ще се роди болно теле. За да се избегнат тези рискове е необходимо броя на хетерозиготните индивиди да бъде намален и при поява на възможност те трябва да бъдат изключени изцяло от разплод. Американската Ангус Асоциация публикува обширен списък на ДНК тестираните животни за липса (AMF- Arthrogryposis Multiplex свободни) или наличие (AMC- Arthrogryposis Multiplex носители) на мутантен AM ген. Този подробен списък осигурява преобладаващ брой от бици с подходящ генотип, които да се използват за изкуствено осеменяване. По този начин търговците на бременни крави могат да осигурят генетично здрави животни за фермерите при минимален риск чрез използване на този списък. Например ако се използва за изкуствено осеменяване син на бик за който е известно, че е носител на мутантния алел то този син има вероятност 50% също да е носител на този алел. Дъщерите на този син, ако останат в стадото има вероятност 25% също да са носители на мутантния алел и чрез тяхното потомство алелът да се разпространи все повече и повече в това стадо. Следователно стадото може да бъде свободно от АМ само ако за изкуствено осеменяване се използват свободни от ген за АМ бици. Това може да се осъществи само чрез използване на ДНК тестирани бици и крави. Ако отидем по-нататък използването на ДНК генотипизирането ще предложи на потенциалните клиенти възможност за информирани решения при покупка на нови бици или крави. Тези разсъждения имат логическо значение за регистрираните чистопородни животни от породата Ангус, но в същия момент те важат и за кръстоските или новите породи получавани с участието на Ангус, тъй като мутантния алел се наследява по същия начин както и при чистопородните Ангуси. В заключение може да се каже, че АМ вече не е голям проблем при търговията с разплодни животни. ДНК тестът е единствения начин за откриване на носителите на мутантния алел и по този начин да се предотврати и изкорени появата на това нанасящо икономически загуби заболяване. На практика ДНК теста се осъществява чрез вземане на кръвни или тъканни проби от животните, които ще се използват за разплод или търговия и изследването им в определените за това лаборатории. Специално за породата Ангус са определени три лаборатории: AGI DNA Tests, Pfizer® HD50K и IGENITY® Profile, където животните може да се тестват и за други наследствени заболявания (виж приложената таблица долу). Допълнителна информация може да се получи от сайта на American Angus Association.
Литература.
1. Scott P. Greiner, Ph.D., Extension Animal Scientist, VA Tech. Livestock Update, March 2009.
Адхезионен дефицит на говеждите лимфоцити (Bovine Lymphocyte Adhesion Deficiency - BLAD). Наследствената болест BLAD е открита при породата Холщайн през осемдесетте години на миналия век и се наследява като автозомно-рецесивен признак. Чрез фамилен анализ е доказано, че тази мутация е разпространена от бика Osborndale Ivanhoe, за който се знае, че е оказал силно влияние върху повишаването на млечната продуктивност на породата. Неговите синове Penstate Ivanhoe Star и Carlin-M Ivanhoe Bell също са хетерозиготни и отговорни за разпространението на болестта. Счита се, че може би 10% от регистрираните крави в САЩ от тази порода са дъщери на тези бици. Според различни съобщения 24% от регистрираните за изкуствено осеменяване бици са носители на мутантния алел, но чрез въвеждане на диагностичното генотипизиране този брой ще бъде значително намален. Молекулната същност на BLAD е точкова мутация (замяна на аденин с гуанин) в позиция 383 на ген CD18, което от своя страна е причина за промяна в кодираната от този ген полипептидна верига чрез замяна на аспартовата киселина с глицин в позиция 128 (D128G) на адхезионната молекула CD18. Открита е и втора точкова мутация, която е тиха (без функция). Неутрофилите на болните от BLAD говеда имат непълна експресия на beta2 интегрина (CD11a,b,c/CD18) на левкоцитната адхезионна молекула. Отклоненията в адхезионните функции на левкоцитите при болните животни са вече напълно описани. Клиничните признаци на това заболяване се проявяват на възраст 1-2 седмици и се характеризират с язви по устната лигавица, тежък периодонтит, липса на зъби, хронична пневмония и повтаряща се хронична диария. Хематологичните изследвания показват неутрофилия (често >100,000) без изместване наляво, лимфоцитоза и моноцитоза. Биохимичните изследвания се характеризират с хипоалбуминемия, хиперглобулинемия, ниски стойности на креатинина, уреята и глюкозата. Болните телета умират на възраст 2-4 месеца поради инфекциозни усложнения. Отделни животни могат да доживеят 1-2 години, но те са недоразвити и със силно влошени стопански качества. Биците от Холщайнската порода се контролират за BLAD в продължение на повече от десетилетие. Контролът върху заболяването се осъществява чрез публикуване на генотипизираните бици и избягване на съешаване между хетерозиготни животни. За диагностика на това опасно заболяване
The Holstein Association of America използва лабораториите Immgen и Genetic Visions, но напоследък използва услугите и на Канадската лаборатория Genetic Analytics. Цената за един тест е около $40.
Литература.
1. Ackermann, M.R., et al. Alimentary and respiratory tract lesions in eight medically fragile Holstein cattle with bovine leukocyte adhesion deficiency. Veterinary Pathology. 33 (3) :273-281 (1996).
2. Arrayet, J.L et al. Growth of Holstein calves from birth to 90 days: The influence of dietary zinc and BLAD status. Journal of Animal Science. 2002. 80:545-552.
3. BLAD. Immgen. October 13, 2004
4. http://www.immgen.com/Services/Traits/BLAD.html.
5. BLAD. http://astronaut.agoff.umn.edu.ansc3221/Blads/ 1999.
6. Holstein Associaton of America, Brattleboro, VT. Telephone conversation, March 15, 2005.
7. Ribeiro, Luciana A., et al. PCR screening and allele frequency estimation of bovine leukocyte adhesion deficiency in Holstein and Gir cattle in Brazil. Genetics and Molecular Biology. 23 (4): 831-834 (2000).
8. Smith, Bradford P. Large Animal Internal Medicine 3rd ed. Mosby, Inc. 2002, рp.1498 and 1603.
9. van Garderen, E. et al. Post-mortem findings in calves suffering from bovine leukocyte adhesion deficiency. Vet Q. 1994 Mar 16 (1): 24-6.
10. Wanner, J.M et al. Clinical Mastitis in Primiparous Holsteins: Comparisons of Bovine Leukocyte Adhesion Deficiency Carriers and Noncarriers. http://jds.fass.org/cgi/content/abstract/82/11/2517.
11. Nagahata H. Bovine leukocyte adhesion deficiency (BLAD): a review. The Journal of Veterinary Medical Science / the Japanese Society of Veterinary Science, 2004, 66(12):1475-82.
Наследствен дефицит на ензима уридин монофосфат синтетаза (Deficiency of Uridine Monophosphate Synthetase – DUMPS). Елиминирането на животните болни от наследствени болести и тези, които са носители на патологични гени е от интерес на всички, които се занимават с отглеждане на животни. Разкриването на молекулните основи на генетичните дефекти дава възможност да се открият техните носители на ниво ДНК и което е по-важно, това може да стане в ранна възраст и дори на ембрионално ниво. Съществуват няколко автозомно-наследяващи се наследствени болести при говедото като например дефицита по уридин монофосфат синтетазата и комплексната вертебрална малформация. Дефицитът по уридин монофосфат синтетазата е наследствено заболяване, което се наследява като автозомно-рецесивен признак и предизвиква при говедото ранна ембрионална смъртност. В клетките на бозайниците уридин монофосфат синтетазата (UMPS) е ензим отговорен за конверсията на оротовата киселина до уридин монофосфат, който от своя страна е важен компонент на пиримидин нуклеотидите. Този ензим всъщност има две ензимни функции: оротова фосфорибозил трансфераза и оротидин монофосфат декарбоксилаза, която кореспондира с последните два етапа на пиримидин синтетазата. Мутацията С-Т (т.е. заместване на цитозин с тимин) в гена кодиращ уридин монофосфат синтетазата при кодон 405 от екзон 5 на хромозома 1 води до образуване на стоп кодон и вследствие на това се образува функционално увреден ензим. Носителите (т.е. хетерозиготните индивиди) са фенотипно здрави, но имат в генома си един нормален и един мутантен алел за уридин монофосфат синтетаза. През време на лактацията тези крави отделят в млякото и урината по-голямо количество оротова киселина. Хомозиготните по мутантния алел ембриони не оцеляват до раждане и обикновено умират през ранен етап на бременоста. Ембрионите вероятно се абортират или реабсорбират около 40-я ден от бремеността и това предизвиква повтарящи се маточни кръвоизливи. Повечето от хетерозиготните индивиди открити в Северна Америка (n= 438) и Европа (n= 314) са потомци на бик Happy Herd Beautician, който е един от петте най-добри бици на породата Холщайн в САЩ през 1987 година поради високия си наследствен потенциал за млечна продуктивност както и някои други показатели. Наличие на мутация в гена кодиращ уридин монофосфат синтетазата е открита още в Аржентина, Унгария, Тайван и Турция. Има съобщения, че заболяването не е открито в Полша и Индия, но според нас това, че до този момент заболяването не е открито не означава, че го няма в тези държави. Пример за това е Турция – първоначално и тя беше в списъка на държавите свободни от заболяването, но колеги от Университета в Анкара съобщиха, че са го открили при крави внесени от САЩ. Много е вероятно дефицитът по уридин монофосфат синтетазата да съществува във всички държави, които са внесли замразена сперма или живи животни от САЩ.
Литература.
1. A. R. Rezaee, M. R. Nassiry, B. Sadeghi, A. Shafagh Motlagh, M. Tahmoorespour and R. Valizadeh. Implication of complex vertebral malformation and deficiency of uridine monophosphate synthase on molecular-based testing in the Iranian Holstein bulls population. African Journal of Biotechnology Vol. 8 (22), pp. 6077-6081, 16 November, 2009
2. Akyuz B, Ertugrul O (2008). Detection of deficiency of Uridine Monophosphate Synthase (DUMPS) in Holstein and native cattle in Turkey. Ankara Univ. Vet. Fak. Derg. 55: 57-60.
3. Boom R, Sol CJA, Salimans MMM, Jansen CJ, Wertheim-Van Dillen PME, Van Der Noordaa J (1990). Rapid and simple method for purification nucleic acid. J. Clin. Microbiol. 28: 495-503.
4. Citek J, Blahova B (2004). Recessive disorders – a serious health hazard. J. Appl. Biomed. 2: 187-194.
5. Citek J, Rehout V, Hajkova J, Pavkova J (2006). Monitoring of the genetic health of cattle in the Czech Republic. Vet. Med. 51: 333-339.
6. Esmaelizad M (2003). Molecular characterization of umps and asas genes and identification carriers of two genetic disorder Dumps and cytrollinaemia in Iran. Razi Vaccine and Serum Research Institute in Iran.
7. Fesus L, Anton I, Zsolnai A (1999). Marker assisted selection in livestock. DUMPS, Weaver-diseases and Citrullinaemia in cattle populations. Allatt.-es- Takarm. 48: 193-203.
8. Harlizius B, Schrober S, Tammen I, Simon D (1996). Isolation of the bovine uridine monophosphate synthase gene to identify the molecular basis of DUMPS in cattle. J. Anim. Breed. Genet. 133: 303-309.
9. Kaminski S, Grzybowski G, Prusak B, Rusc A (2005). No incidence of DUMPS carriers in Polish dairy cattle. J. Appl. Genet. 46(4): 395-397.
10. Lee YK, Chang KW, Nam IS, Chang WK, Tak TY, Kim KN, Lee KJ (2002). Studies on the detection of congenital genetic disorder in Holstein proven and candidate bulls. J. Anim. Sci. Technol. 44: 279-288.
11. Lin DY, Huang YC, Chang HL, Liaw RB, Lee SC, Chen JC, Wu SC, Wu MC (2001). DNA typing of in- herited Deficiency of Uridine Monophosphate Synthase in dairy cattle and beef cattle. J. Chin. Soc. Anim. Sci. 30: 15-22.
12. Nassiry MR, Norouzy A, Eftekhari Shahroudi F, Javadmanesh A, Alishad M (2005). Investigation of two recessive disorders in breeder bulls of Abbas Abad animal breeding center. Iranian J. Biotechnol. 3(2): 125-128.
13. Norouzy A, Nassiry MR, Eftekhari Shahroudi F, Javadmanesh A, Mohammad Abadi M, Sulimova G (2005). Identification of Bovine Leucocyte Adhesion Deficiency (BLAD) Carriers in Holstein and Brown Swiss AI Bulls in Iran. Russian J. Genet. 41(12): 1409-1413.
14. Patel RK, Singh KM, Soni KJ, Chauhan JB, Sambasiva Rao KRS (2006). Lack of carriers of citrullinaemia and Dumps in Indian Holstein cattle. J. Appl. Genet. 47(3): 239-242.
15. Poli MA, Dewey R, Semorile L, Lozano ME, Albarino CG, Romanowski V, Grau O (1996). PCR screening for carriers of bovine leukocyte adhesion deficiency (BLAD) and uridine monophosphate synthase (DUMPS) in Argentine Holstein cattle. Zentralbl Vet. A43: 163-168.
16. Robinson JL, Popp RG, Shanks RD, Oosterhof A, Weerkamp JH (1993). Testing for Deficiency of Uridine Monophosphate Synthase among Holstein- Friesian cattle of North America and Europe. Livest Prod. Sci. 36: 287-298.
17. Rusc A, Kaminski S (2007). Prevalence of complex vertebral malformation carriers among Polish Holstein-Friesian bulls. J. Appl. Genet. 48(3): 247-252.
18. Schwenger B, Schober S, Simon D (1993). DUMPS cattle carry a point mutation in the Uridine Monophosphate synthase gene. Genomics, 16: 241-244.
19. Steffen D (2001). CVM threatens Holstein herds. Vet. Pract. News: 36:p. 27.
20. Thomsen B, Horn P, Panitz F, Bendixen E, Petersen AH, Holm L (2006). A missense mutation in the bovine SLC35A3 gene, encoding a UDP-N- acetylglucosamine transporter, causes complex vertebral malformation. Genome Res. 16: 97-105.
Комплескна вертебрална малформация (Сomplex vertebral malformation - CVM). Комплескната вертебрална малформация (CVM) е наследствен летален синдром при телетата от породата Холщайн, който се наследява като автозомно-рецесивен признак и се характеризира с деформирани прешлени в шийната, торакалната и лумбалната област на на гръбначния стълб, аномалии в стернума и ребрата, ниско живо тегло и латерална ротация на ставите в метакарпалната и метатарзалната област. При изследване на сърцето се установява дясностранна хипертрофия, дефект в преградата между предсърдията и много фетуси с CVM се абортират на 159-я ден от бремеността, а други се раждат преждевременно или мъртви. Молекулната основа на CVM е заместване на гуанин с тимин (G-T) в ген 3 (SLC35A3), кодон 559 на генна фамилия 35, хромозома 3, който кодира UDP-N-ацетилглюкозамин транспортера. Тази трансверсия предизвиква заместване на аминокиселината валин с фенилаланин в позиция 180 на полипептидната верига. Основният предшественик на говедата с тази мутация е бик Carlin-M Ivanhoe Bell, който е бил един от елитните бици в породата Холщайн-Фризийско говедо и е бил роден през 1974 година. Най-напред CVM е открита при телета от породата Датски Холщайн през 2000 година и след това наличието на CVM носители са докладвани и в други държави като Швеция, Дания, Чехия и Полша. Поради това, че замразена сперма от бици Холщайн е използвана интензивно и за кросбредни програми с местни говеда е необходимо да се направи скрийнинг на всички кросбредни животни и особено на биците - кръстоски, които се използват за изкуствено осеменяване за да се намали риска от разпространение на това наследствено заболяване.
Литература.
1. Agerholm J. S., Ch. Bendixen, J. Arnbjerg, O. Andersen. Morphological variation of ‘‘complex vertebral malformation’’ in Holstein calves. J. Vet. Diagn .Invest. 16:548–553 (2004).
2. Agerholm JS, Bendixen C, Andersen O, Arnbjerg J (2001). Complex vertebral malformation in Holstein calves. J. Vet. Diagn. Invest. 13: 283-289.
3. Agerholm JS, Andersen O, Almskou MB, Bendixen C, Arnbjerg J, Aamand GP (2004). Evaluation of the inheritance of the complex vertebral malformation syndrome by breeding studies. Acta Vet. Scand. 45: 133-137.
4. Berglund B, Persson A, Stalhammar H (2004). Effects of complex vertebral malformation on fertility in Swedish holstein cattle. Acta Vet. Scand. 45: 161-165.
5. Duncan RB, Carrig CB, Agerholm JS, Bendixen C (2001). Complex vertebral malformation in a Holstein calf : report of a case in the USA. J. Vet. Diagn. Invest. 13: 333-336.
6. Nagahata H, Oota H, Nitanai A, Oikawa S, Higuchi H, Nakade T, Kurosawa T, Morita M, Ogawa H (2002). Complex Vertebral Malformation in a Stillborn Holstein Calf in Japan. J. Vet. Med. Sci. 64(12): 1107-1112.
7. Nielsen US, Aamand GP, Andersen O, Bendixen C, Nielsen VH, Agerholm JS (2003). Effects of complex vertebral malformation on fertility traits in Holstein cattle. Livest Prod. Sci. 79: 233-238.
8. Revell S (2001). Complex vertebral malformations in a Holstein calf in the UK. Vet. Rec. 149: 659-660. Robinson J, Shanks RD (1990). Deficiency of uridine monophosphate
synthase among Holstein cattle. C. Vet. 80: 119-122.
Наследствен дефицит на фактор XI (Factor XI – plasma thromboplastin antecedent deficiency in cattle). Сега вече е известно, че по-голямата част от животинските видове принадлежащи към клас Mammalia (бозайници) имат в известна степен подобен коагулационен механизъм, но нивото на активност на съсирващите фактори варира в зависимост от вида животни. В специализираната литература са описан наследствени дефицити при почти всички известни коагулационни фактори. Обаче с изключение на кучето има сравнително малко научни съобщения за наследствени коагулационни дефекти при другите видове животни. При говедото са описани случаи на придобити кръвозливи след изхранване на развалена детелина и орлова папрат, но има само едно съобщение за наследствен коагулационен дефицит. Kociba et al. (1969) съобщават за наследствен дефицит на предшественика на плазмения тромбопластин (Factor XI, PTA) при две кастрирани животни от породата Холщайн. Резултатите от изследването на животните показват, че този дефицит вероятно е същия както при човека и протича с умерена клинична манифестация. Преди кастрацията им телетата са изследвани и е установено удължено време за коагулация на кръвта. Едното животно е кастрирано без да показва проблеми с кръвосъсирването, но второ биче е умряло от травматичен ретикулит преди да завърши изследването за коагулация на кръвта.
Сравнително изследване на различни коагулационни параметри при говеда Холщайн
Нормални стойности |
Стойности при кастратите |
|
Време за съсирване (mins) |
||
(a) в стъклени епруветки |
6-20 |
55 |
(b) в силиконови епруветки |
12-60 |
95 |
Едноетапно протромбиново време (sec) |
22-28 |
22 |
Изследване с отрова на пепелянка на Ръсел (sec) |
21-51 |
21 |
Частично тромбопластиново време (sec) |
46-52 |
308 |
Протромбиново време (sec) |
24-102 |
22 |
Фибриноген (mg per 100 ml) |
680-820 |
728 |
Тромбоцити брой в mm3 (x 103) |
200-600 |
570 |
Активност на специфичните фактори за кръвосъсирване при кастратите
(в % от нормата) |
|
Протромбин (Фактор II) |
114% |
Проакцелерин (Фактор V) |
108% |
Фактор VII |
100% |
Антихемофилен фактор (Фактор VIII) |
100% |
Фактор на Христос (Фактор IX) |
107% |
Фактор на Стюарт (Фактор X) |
104% |
Предшественик на плазмения тромбопластин (Фактор XI) |
1% |
Фактор на Хагеман (Фактор XII) |
92% |
Фибрин стабилизиращ фактор (Фактор XIII) |
100% |
Анти фактор XIa |
74% |
Gentry и Black (1980) изследват 169 Холщайн-Фризийски говеда и откриват 21 хетерозиготни по фактор XI животни, които са имали средна активност на този фактор 0,43 ± 0,10 единици на милилитър при 0,99 ± 0,32 единици на милилитър. Честотата на мутантният ген е била 0,06, а честотата на откритите хетерозигтно генотипове – 0,12. Проведеният фамилен анализ е показал, че това наследствено заболяване се наследява като автозомно-рецесивен признак.
През 1994 г. Gentry и Ross (4) продължават изследванията си върху фактор XI при Холщайнски крави и откриват, че 47 от тях са хетерозиготни и една е хомозиготна по мутантния алел. Тези изследвания показват, че честотата на носителите на този алел се е повишила значително и че това изизсква сериозна борба с разпространението му. За целта е необходимо да се извърши масов скрийнинг на първо време само при биците определени за изкуствено осеменявани и майките-бикопроизводителки.
Patel et al. (2007) изследват в Индия 1001 животни от видовете Bos taurus и като представител на вида е избрана породите Холщайн и Джерсей, Bos indicus, кръстоски B. taurus x B. indicus и речни биволи без да се уточнява породата им. Открити са два хетерозиготни бика от породата Холщайн, а останалите животни са били свободни от мутацията. Извършеният сравнителен секуенсинг между нормални и хетерозиготни животни е показал наличието на инсерция с размер 77 чифта бази (AT (A)29 TAAAG (A)27 GAATTATTAATTCT) в рамките на екзон 12 на ген FXI.
В съседна Турция при изследване на 350 Холщайнски крави Meydan et al. (2010) откриват четири хетерозиготни животни (1,2%), което е голямо указание за наличието на това заболяване.
За САЩ се използва ДНК тест предложен от Marron et al. (2004) и чрез него е намерено, че честотата на мутантния алел е 1,2% от биците Холщайн през посочената година.
В България не е известно да е извършвано такова изследване, но е извършван многократен внос на живи животни Холщайн и замразен семенен материал от САЩ и други държави, което ни дава основание да предполагаме, че заболяването го има и у нас.
Литература.
1. KOCIBA, G. J., О. D. RATNOFF, W. F. LOEB, R. L. WALL and L. E. HEIDER. Bovine
plasma thromboplastin antecedent (Factor XI) deficiency. J. Lab. clin. Med., 74: 37-41, 1969.
2. Gentry P. A., S. Crane and F. Lotz. FACTOR Xi (PLASMA THROMBOPLASTIN ANTECEDENT) DEFICIENCY IN CATTLE. CAN. VET. JOUR., 16, 6, 160-163, 1975.
3. Gentry P. A., W.D. Black. Prevalence and Inheritance of Factor XI (Plasma Thromboplastin Antecedent) Deficiency in Cattle. Journal of Dairy Science, 63, 4, 616-620, 1980.
4. Gentry P. A. and M. L. Ross. Coagulation Factor XI Deficiency in Holstein Cattle: Expression and Distribution of Factor XI Activity. Can. J. Vet. Res. 1994; 57: 242-247.
5. Marron B. M., J. L. Robinson, P. A. Gentry, J. E. Beever. Identification of a mutation associated with factor XI deficiency in Holstein cattle. Animal Genetics, 35, 6, 454–456, 2004.
6. Meydan H., M. A. Yildiz1, J. S. Agerholm. Screening for bovine leukocyte adhesion
deficiency, deficiency of uridine monophosphate synthase, complex vertebral malformation, bovine citrullinaemia, and factor XI deficiency in Holstein cows reared in Turkey. Acta Veterinaria Scandinavica 2010, 52:56, 2-8.
Наследствена тромбопатия при породата Симентал (Platelet Bleeding Disorder, Simental hereditary thrombopathy). За първи път наследствено обусловената неспособност на тромбоцитите да се агрегират е описана през 1980 г. от Kociba при породата Симентал. От тогава до сега са направени още много съобщения за това заболяване. Интересното, е че засега заболяването е установено само при Симентала. Най-много болни животни са открити в САЩ и Канада. В клинично отношение заболяването се характеризира с спонтанен епистаксис (кръвотечение от носа), повърхностни хематоми, продължителни кръвотечения при нараняване или дори най-леки манипулации като татуиране или инжекции. Наблюдавани са също спонтанни мукозни кръвоизливи и хематурия. Наблюдавани са тежки кръвоизливи от носа особено при студено време. При по-сериозни хирургически интервенции, особено в коремната област са налюдавани профузни кръвоизливи. Животните са анемични и слаби. Компенсаторната еритропоеза е нарушена поради значителното намаляване на желязото вследствие на хроничните кръвотечения. Броят и морфологията на тромбоцитите не са нарушени. Агрегационната им способност определена ин витро е силно нарушена. Това всъщност е основната причина за кръвоизливите при болните животни и този дефект е установен от всички изследователи, които са работили върху този проблем. При аутопсия трудно се установява къде най-напред е започнало кръвотечението. Типичен случай за това е крава с пулмонална хеморагия и епистаксис. Steficek et al. (1993) предполагат, че заболяването има наследствен характер и поради това го наричат Наследствена тормбопатия при Симентала (Simmental hereditary thrombopathy). Mapletoft et al. (2000) доказват наследствения характер на заболяването чрез използване на технологията за ембриотрансфер за да се получат повече потомци от хетерозиготните родители. По този начин са получени 27 телета от които 25 са били фенотипно здрави, а две болни т.е. те са имали силно нарушена способност на тромбоцитите да се агрегират. Според тези автори заболяването не се наследява като прост менделиращ признак, а се детерминира най-малко от два гена. През 2007 Boudreaux et al. разкриха молекулната същност на това наследствено заболяване. Чрез секвениране на гена, кодиращ CalDAG-GEFI (calcium diacylglycerol guanine nucleotide exchange factor I) при здрави и болни телета те откриват, че причината за заболяването е нуклеотидна замяна Т-С в позиция 701, което от своя страна води до заместване на аминокиселината пролин с левцин, а това е причина за структорна промяна във втори регион (SCR2) на каталитичния домейн на специфичния протеин отговорен за агрегацията на тромбоцитите. Според авторите тази точкова мутация е отговорна за тромбопатията при Симентала. Наследствената тромбопатия при Симентала е много подобна на тромбопатията при породите кучета Басет хаунд и Ескимо шпиц (тази тромбопатия при кучетата е подробно описана от Сотиров, 2011). При тези две породи кучета са намерени две различини точкови мутации в гена, кодиращ CalDAG-GEFI, а при породата Ландзеер (Landseer) от Европейски тип е установена и трета мутация. При кучетата окончателно е прието, че заболяването се наследява като автозомно-рецесивен признак въпреки установените повече от една точкови мутации и поради това може да се приеме, че при породата Симентал наследствената тромбопатия се наследява по същия начин. Между впрочем методът за ДНК диагностика на Наследствената тромбопатия при Симентала предложен от Boudreaux et al. (2007) е одобрен за използване в САЩ.

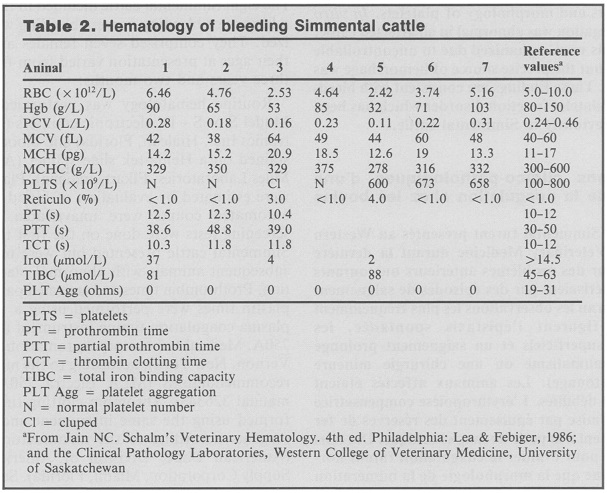
Патологоанатомичната находка извършена върху три евтаназирани животни е показала тежки кръвоизливи в абомазуса, цекума, подкожни и интрамускулни хеморагии, кръвоизливи в червата и перитонеума. Животните са били евтаназирани поради неконтролируем кръвоизлив от ноздрите и изтичане на кръв в трахеята и бронхите. При едно от животните са намерени обширни рекурентни хематоми в областта на лявата скапула и левия хълбок. Намерени са още множествени кръвонасядания по задните крайници и дясната подмишница. Илиачните и сублумбалните лимфни възли са били жълто-кафяви. Хистологичното изследване на тези лимфни възли е показало изпълване на трабекуларните и медуларните синуси с макрофаги погълнали голямо количество хемосидерин.
Литература.
1. BOUDREAUX M. K., S. M. SCHMUTZ, AND P. S. FRENCH. Calcium Diacylglycerol Guanine Nucleotide Exchange Factor I (CalDAG-GEFI) Gene Mutations in a Thrombopathic Simmental Calf. Vet. Pathol., 2007, 44: 932- 935.
2. Dodds W.J. Hemostasis. In: Kaneko JJ, ed. Clinical Biochemistry of Domestic Animals. 4th ed. Toronto: Academic Press, 1989: 274-315.
3. Kociba G.J. Partial characterization of a hemostatic defect in Simmental cattle. Vet. Clin. Pathol., 1980; 9: 46-47.
4. Leipold H., Moore W. Bovine Thrombopathia. Listed by Dodds WJ, in First Registry of Animal Models of Thrombosis and Hemorrhagic Diseases. ILAR News 1977; 21: A14.
5. Mapletoft R. J., Sh. M. Schmutz, G. P. Searcy. A study of the inheritance of a bleeding disorder in Simmental cattle. Can. Vet. J., 41, 791-793, 2000.
6. Persijn J.P., Van Der Silk W., Riethorst A. Determination of serum iron and latent iron-binding capacity (LIBC). Clin. Chim. Acta., 1971; 35: 91-98.
7. Searcy G. P., L. Petrie. Clinical and laboratory findings of a bleeding disorder in eight Simmental cattle. Can. Vet. J., 1990; 31: 101-103.
8. Searcy G. P., D. Sheridan and K. A. Dobson. Preliminary Studies of a Platelet Function Disorder in Simmental Cattle. Can. J. Vet. Res., 1990; 54: 394-396.
9. Steficek B. A., J. S. Thomas, J. C. Baker, T. G. Bell. Hemorrhagic diathesis associated with a hereditary platelet disorder in Simmental cattle. J. Vet. Diagn. Invest., 5:202-207 (1993).
10. Сотиров Л. Наследствени болести при кучетата, 2011, Стара Загора, Кота, под ред. на Х. Хубенов и М. Паскалев.
Протопорфирия (Protoporphyria, Congenital Erythropoietic Porphyria, Pink tooth, Osteohemochromatosis). Порфирията е описана за първи път през 1874 г. при човека от Sohultz и Baumstark и при кравите от Bronvеr през 1883 г. От тогава са описани и други случаи при говеда и свине, които са се появявали при клане на такива животни и са показвали кафеникаво оцветяване на костите и червеникав цвят на зъбите. През 1910 г. е разкрита порфириновата същност на описаното пигментиране на костите и зъбите от Dane Valdemar Pulsen в нейната класическа монография и на заболяването е дадено оригиналното наименование “Osteohemochromatosis”. Заболяването е известно при животните главно от материал взет пост мортем, докато Fourie et al. (1936-1939) не го описват и при живи говеда в Южна Африка. Установено е и при живи прасета от Clare and Stephens (1944) в Нова Зеландия. Порфирията е рядко заболяване при говедата и свинете и затова е било голяма изненада едновременото му появяване в графство Тистед, северна Дания през годините 1951-1954. Прасетата са били открити при клане в Тистед през 1954 г. и Jorgensen (1959) доказва, че те са потомци на един нерез, в който вероятно се е появила мутацията. Jorgensen запазва няколко прасета за по-задълбочени изследвания заедно с други изследователи (With et at.,1959) в Селскостопанската експериментална станция, Копенхаген. За съжаление болните от порфирия прасета са измрели, най-вероятно от допуснатия високостепенен инбридинг. В посоченото графство родителите на болните прасета са били отстранени от разплод и така постепенно популацията на местните свине е била освободена от мутантния ген.
Както се каза Протопорфирията е рядко наследствено заболяване при говедата, свинете, котките и човека, при които се образува дефектен хемоглобин в резултат на голямо образуване на Тип I порфирини в ядрата на образуващите са нормобласти. Този дефект при говедата се наследява като автозомно-рецесивен признак и обикновено се ограничава в стадата, където се допуска родствено или затворено линейно развъждане. Заболяването е установено в САЩ (най-напред през 1977 г.), Франция (1991)Канада, Дания, Ямайка, Англия, Южна Африка, Австралия (1992) и Аржентина. Преди 2077 г. заболяването е рядко съобщавано в Обединеното кралство, но когато от лятото на същата година започват масови изследвания на телетата от породата Лимузин са открити много случаи особено в Англия и Уелс. Това голямо географско разпространение показва, че заболяването го има по целия свят и вероятно засяга всички животни с месодайна насоченост и особено говедата, свинете и овцете. Хетерозиготните животни са здрави, но хомозиготните по рецесивния алел показват признаци на заболяването още при раждане – червеникави на цвят зъби, кости и урина, които перзистират през целия живот на болните животни. Наследственият ензимен дефект е причина за дефицит в активността на ензима уропорфириноген III косинтетаза, която е важна част от порфирин-хем биосинтезата. Урината на засегнатите животни съдържа високи количества на копротопорфирин I и уропорфирин I и поради това цвета й е кехлибарен или червено-кафяв. Костите, урината и зъбите (особено млечните зъби) флуоресцират розово, когато се облъчат с ултравиолетова светлина отблизо. Продължителното излагане на слънчева светлина, поради фотосензибилизацията, причинява типични лезии с хиперемия, образуване на везикули и повърхностни некрози на непигментираните участъци на кожата. Тежестта на кожните лезии зависят от интензивността на слънчевата радиация и степента на кожната пигментация, която се наблюдава при някои фамилии животни. Развива се нормохромна хемолитична анемия с увеличен брой на макро и микроцити и точковидно оцветяване на базофилите. Понякога се наблюдава и спленомегалия. Структурата на костите не е променена с изключение на случаите при които костите имат повишена чупливост, дължаща се на изтънен кортекс. Засегнатите животни обикновено са в добро състояние докато не настъпят описаните кожни изменения. Някои животни растат добре докато се пазят от слънчева светлина. Някои форми на протопорфирията причиняват фотосензибилизация само при говедата от породата Лимузин и човека. При човека е описана цяла серия от различни порфирии, които се дължат на увредени функции на ензимите, които осъществяват порфирин-хем биосинтезата и са групирани в зависимост от клиничните признаци. Те варират широко от тежки кожни лезии на откритите части на тялото, остри фотосенситивни реакции, сериозни увреждания на черния дроб и остри атаки на нервната система. При животните тези заболявания са класифицирани в три форми – конгенитална еритропоетична порфирия, конгенитална еритропоетична протопорфирия и порфирия. Вероятно всички синдроми описани при човека съществуват и при животните и поради това описаната класификация може да бъде разширена. Това наследствено заболяване при прасетата и котките е много рядко и се различава от това при говедата с това, че липсва фотосензибилизация. При тях заболяването се наследява като автозомно-доминантен признак. При прасетата, дори с високи нива на порфирини в кръвта, фотодерматитен ефект няма. Диагнозота се базира върху екскрецията на анормални уропорфирини, червеникаво оцветяване на зъбите, които флуоресцират розово, когато се облъчат с ултравиолетова светлина, оцветена с кехлибарен или червено-кафяв цвят урина и хемолитична анемия. Концентрацията на общия плазмен порфирин е силно повишена (норма <10nmol/l; повишение до 100 nmol/l). Общият еритроцитен порфирин показва подобно увеличение. При хистологично изследване се открива характерно отлагане на пигмент подобен на липофусцина в хепатоцитите и Купферовите клетки на черния дроб. Някои отлагания имат характерна форма на Малтийски кръст при наблюдаване с поляризирана светлина.
Диференциална диагоноза:
1. Първична фотосензибилизация, дължаща се на приемане на фотодинамични агенти като хиперицин чрез растението Жълт кантарион (Hypericum perforatum).
2. Вторична фотосензибилизация при тежко чернодробно увреждане от високи нива на фотодинамичния агент филоеритрин (образува се от хлорофил).
3. Малигнена катарална треска (Malignant catarrhal fever).
4. Син език (Bluetongue - BTV-8).
5. Когенитална порфирия при други породи говеда като Шортхорн, Айршир и Фризийско говедо, коята се наследява също автозомно-рецесивно и има същите клинични признаци.
Носителството на рецесивния алел е широко разпространено при говедата, но хомозиготните по него животни са сравнително малко. Това е свързано с характера на това наследяване. Хетерозиготите са клинично здрави и имат по-ниски нива на уропорфириноген III косинтетаза в сравнение с генотипно здравите животни. Лабораторната биохимична идентификация е възможно да се прави, но рядко се прилага поради относително малкото инциденти. Това разбира се е сериозен пропуск, защото може да доведе до повишаване честотата на хетерозиготните индивиди и да се стигне до “взрив” на заболяването при насищане с тях на популациите, особено на тези с малък брой. Смъртността на болните животни може да се контролира чрез предпазването им от пряка слънчева светлина особено през месеците с висока слънчева активност.
Понастоящем The North American Limousin Foundation (NALF), Canadian Limousin Foundation и Australian Limousin Breeders Association използват PCR метод за ранна диагноза и откриване на носителите на мутантния алел. Той се основава на амплификация на мутантния ферочелетазен ген. В САЩ за да се запише новото поколение в племенната книга на породата, биците определени за изкуствено осеменяване трябва да са генотипизирани за протопрфирия. В Обединеното кралство такава обаче засега няма.
Литература.
1. Jenkins M.M., LeBoeuf R.D., Ruth G.R., Bloomer J.R.: A novel stop codon mutation (X417L) of the ferrochelatase gene in bovine protoporphyria, a natural animal model of the human disease. Biochim. Biophys. Acta., 1998, 1408:18–24.
2. Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. Whitehouse Station NJ, USA, 2011.
Прогресивна дегенеративна миелоенцефалопатия при говедото (Bovine progressive degenerative myeloencephalopathy - BPDME, weaver syndrome). Това е нервно-дегенеративно заболяване при Кафявото Швейцарско говедо (още се нарича Сивокафяво алпийско говедо), което е установено в САЩ, Канада и Европа. За да се постави такава диагноза трябва да са задължително налице четири критерия: 1) едновременна билатерална атаксия на задните крайници и проявена дисметрия (неспособност или нарушена способност за точен контрол на степента на движенията при мускулни действия. Също невъзможност за точно определяне на разстояния), когато животните са на възраст 5-8 месеца; 2) неадекватни проприоцептивни отговори (това са рецептори, които се намират главно в мускулите, сухожилията, ставите и вътрешното ухо, които определят движенията или позицията на тялото и крайниците при отговор на дразнене възникнало вътре в организма), атаксия при четирите крайника и прогресивна парапареза; 3) нормални спинални рефлекси и запазена функция на краниалните нерви и липса на тежка мускулна атрофия; 4) фамилна връзка на разпространение при установяване на посочените признаци. Първоначално заболяването се е наричало “преплетена походка” (weaver) поради особеното преплитане на краката при ходене на животното. Хистопатологичните промени са основно в сенсорната нервна система в противоположност на спиналната мускулна атрофия. Спиналната дисмиелинизация причинява конгенитално латерално залежаване и опистотонус, но спиналните рефлекси и съзнанието са нормални.
Прогресивната дегенеративна миелоенцефалопатия при говедото (BPDME) е описано при чистопородните Кафяви швейцарски говеда и при двата пола. Установена е в САЩ, Дания, Германи и Швейцария. Само през 1987 г. са открити над 215 случая само в САЩ. В Дания заболяването е открито при юници кръстоски между Кафяво швейцарско говедо с Червено датско говедо. Кръстоските са получени след използване на замразена сперма от Кафяви американски бици внесена от САЩ. В Германия е описан случай на прогресивна атаксия и дискоординация на задните крайници при 1,5 годишна бременна юница от породата Англер, която е показвала признаци идентични с BPDME. Важно е да припомним, че Кафявото швейцарско говедо е използвано при създаването на породата Англер в Германия и така това заболяване е пренесено в тази порода. Не е установено специфично географско разпространение на BPDME. Най-характерният признак на заболяването е билатерална слабост на задните крайници, когато животните са прави или се опитват да станат след лежане. Първоначално атаксията е слаба, но тя става по-очевидна след по-силни движения на животното. Тези първи признаци на BPDME се проявяват обикновено на възраст между 5 и 8 месеца. През следващите 12-18 месеца болните животни проявяват прогресираща атаксия с прогресираща деориентация на локомоторната функция, която засяга най-напред задните крайници. На възраст 18-24 месеца повечето болни животни развиват тежка атаксия на задните крайници с явно отслабени проприоцептивни рефлекси. През по-късните стадии на заболяването проприоцептивния дефицит става още по-очевиден дори когато животните са прави. Слепите животни болни от BPDME показват по-силно изразена атаксия, дисметрия, проприоцептивен дефицит и люлеещи се задни крайници, когато са в покой. При тежките случаи се забелязва умерена и генерализирана загуба на телесна маса с умерена атрофия на бедрената мускулатура. Не се забелязват нарушения в инервацията на главата. Евентуално животните с тежка атаксия и слабост на задните крайници може да се залежат и може да умрат от вторични усложнения като тимпания наример. Установено е фамилно разпространение на заболяването при Кафявото швейцарско говедо. При едно изследване извършено върху 32 болни животни 48,60% са били потомци на три бика. По-нататъшни изследвания на болните животни са показали, че най-вероятно заболяването се наследява като автозомно-рецесивен признак. Популационното изследване е показало относително висока честота на разпространение на заболяването в породата Кафяво швейцарско говедо (0,56%). В резултат на тези изследвания осем бика са регистрирани в Board of Directors of the Brown Swiss Cattle Breeders Association of the U.S.A. като носители на рецесивния алел (т.е. тези животни са били хетерозиготни). Преди създаването на ДНК тест за диагностика на BPDME за да се обяви един бик за носител на рецесивния алел е било необходимо от него да се получат два или повече болни потомъка и тези бици са били записвани в племенните книги с буквата W като интегрална част от името на бика. Според най-нови генетични изследвания BPDME е свързано с по-висока млечна продуктивност. Тази връзка би могла да се повлияе от плейотропното действие на един ген (рецесивния алел) или генетичното равновесие между гена причиняващ заболяването и QTL локуса за млечна продуктивност. За да се провери тази хипотеза е извършено проучване върху едно родословие за наличие на връзка между микросателитния локус TGLA116 и гена за заболяването (zma, 8.15; 9, 0.03), които са тясно свързани. TGLA116 микросателитния локус и гена за заболяването (zma, 8.15; 9, 0.03) са отбелязани в една синтенна група 13 (т.е. наличие на два или повече гена в една и съща хромозома). Този микросателит може да се използва за да се откриват носителите на рецесивния алел и да се изследва влиянието на кореспондиращия хромозомен регион върху млечната продуктивност при Кафявото швейцарско говедо и други говежди породи.
Литература.
1. MICHEL GEORGES, ALLAN B. DIETZ, ANURADHA MISHRA, DAHLIA NIELSEN, LESLIE S. SARGEANT, ANITA SORENSEN, MICHAEL R. STEELE, XUYUN ZHAO, HORST LEIPOLD, JAMES E. WOMACK AND MARK LATHROP. Microsatellite mapping of the gene causing weaver disease in cattle will allow the study of an associated quantitative trait locus. Proc. Nati. Acad. Sci. USA, Vol. 90, pp. 1058-1062, 1993.
2. John D. Baird, Ulla M. Sarmiento and Parvathi K. Basrur. Bovine Progressive Degenerative Myeloencephalopathy ("Weaver Syndrome") in Brown Swiss Cattle in Canada:
A Literature Review and Case Report. Can Vet J, 29, 370-377, 1983.
3. Braun U, Ehrensperger F, Bracher V. The Weaver syndrome in cattle. Clinical, biochemical and pathologico-anatomic studies in a Braunvieh/Brown Swiss cow with bovine progressive degenerative myeloencephalopathy. Tierarztl Prax. 1987;15(2):139-44.
Джуджевидност при говедата за месо (Dwarfism). Джуджевидните форми са наблюдавани при хората и животните от векове. Много от тези форми се наследяват. Някои учени вярват, че някои породи животни и домашни любимци може да се използват за пропагандирането на тези форми. И наистина едни от най-ранните съобщения за джуджевидност са направени при говедото. Такива джуджевидни форми е описал в своя дневник Чарлз Дарви още през 1883 г. при своите посещения в Южна Америка: “При два случая аз срещнах в тази провинция няколко вола от много куриозна порода наречена Ната или Ниата. Те външно приличаха на другите говеда така както бултериера и пуг приличат на другите кучета. Предните им крайници са много къси и широкопоставени, върха на носа е извит нагоре, а горната устна е обърната назад, долната им челюст покрива горната по една и съща крива линия и зъбите им са винаги открити. Ноздрите им са поставени високо и са много широки, а очите им са изпъкнали. Когато вървят те държат главата си ниско и тя е свързана с тялото с къса шия, а задните им крака са доста по-дълги от предните. Оголените им зъби, къси глави и обърнати назад ноздри им придават много смешно и самоуверено изражение, което изразява незачитане каквото не можем да си представим”. Когато Дарвин е описвал тези породи е бил на 80 - 90 години. Има изключително сходство на това описание и тези животни, които в тази страна нежно наричат “изключителни” джуджета.
Форми на джуджевидност. Най-често задавания въпрос от развъдчиците, които не харесват джуджетат е – как изглеждат те? На този въпрос не може да се даде един отговор, защото известните форми на джужевидност варират изключително много. Повечето типове, които съществуват в говеждите популации могат да се класифицират в три големи групи. Това групиране се основава на на външния им вид, който е намерен при изследванията извършени в област Манитоба, а също и изследванията на други учени.
(a) Изключителни ("Snorters") – това наименование не винаги се използва от генетиците и физиолозите, но много често се използва от развъдчиците поради трудното и шумно дишане на тези животни. Те се раждат къси и компактни. Предните им крайници са ненормално къси. Главите им може да бъдат твърде квадратни, а долната челюст може да е леко изпъкнала. Предните им крайници често са бъчвообразни. Езикът обикновено се подава от устата, а очите са изпъкнали. Някои или всички тези признаци може да липсват при някои новородени телета. В действителност при чудатите ("snorter") джуджета главата е доста дълга и понякога тясна. Мъртвите раждания са често явление особено при първескините юници, които може да имат и трудно раждане. Често живородените телета не нарастват и имат трудна координация поради късото тяло или твърде свитите сухожилия и нарушено равновесие. Смъртта настъпва често в първите дни след раждане при тези същества. В случаите, когато те оцеляват през периода на растеж разликите между телетата-джуджета и нормалните телета стават очевидни. Те заемат особена поза, корема се увеличава, дишането се затруднява и настъпва общо подуване на тялото. Краката изтъняват особено след отбиване. Скъсяването на крайниците и удебеляването на тялото са непропорционални, а главата е твърде голяма за тялото. На възраст около 3 години те достигат около 1/3 дo 2/3 от нормалното тегло и изглеждат твърде стари. Те обикновено изглеждат като кретени в сравнение с нормалните говеда. Това е нещо подобно на жертвите на синдрома на Hurler при човека. Понякога се наблюдава модифицирана версия на тази форма, при която някои от гротескните признаци липсват. Тази форма най-често е наблюдавана при породата Херефорд, а в Канада най-често се среща при Ангуса. В САЩ има съобщения и за породата Шортхорн. Ниски и набити телета ("Stumpy") са описани от Baker в Небраска и вероятно това е също подтип на тази форма.
(b) Компактна (компресирана) форма – тази форма се среща най-често при Шортхорна в САЩ и се счита, че има фамилно разпространение в някои стада. Животните изглеждат почти нормални, но главата, тялото, шията и краката са леко скъсени в сравнение с нормалните животни. Те се развиват сравнително добре до едногодишна възраст и изглеждат нормални, но като възрастни имат едва 2/3 от нормалните размери. “Компресираните” говеда при Херефорда не са джуджета, но се счита, че са “носители” на мутантния ген и от тях се получават джуджета подобни на "snorter" при съешаването им. Те са и по нежизнени. Счита се, че “Компресираните” животни са потомци на един бик, Colorado Domino 68th, но подобни телета са получени и от други бици, които не са родственици на посочения бик. Това показва, че при тази порода има друг бик от който е разпросранен мутантния алел.
(c) Тип дребосък, дългоглави телета (Miscellany "Longheads", "Pin-heads", Midgets) – тези животни са с нормални пропорции на тялото, но са по-дребни и нямат особено стопанско значение. Дългоглавите телета растат бавно и достигат по-малки размери. Тялото им е удължено и с дълга глава, имат красива муцуна и понякога краката им са криви. Те определено не се развиват добре, но пост мортем при аутопсия не се откриват патологоанатомични изменения. Тази форма е дикусионна, защото някои телета може да приличат повече на описаните по-горе форми. Други джуджевидни форми описани от изследователите са “булдог” (при Декстера и Холщайна) и “патешки крака ("duck legged") при породата Джерсей.
Наследяване. Според Olson (2008) различните форми на джуджевидност се наследяват по автозомно-рецесивен начин. Mishra et al. (2004) установяват, че алелите които причиняват тези дефекти са мутирали по различен начин при различните породи говеда. Например мутацията в ген limbin при Японското кафяво говедо се дължи на нуклеотидно заместване С-Т в позиция 1356 докато при Ангуса мутацията е C-А в позиция 2054/2055. Джуджевидният дефект е установен също и при зебу от породата Браман.
Икономически проблеми. Безпокойството от описания генетичен дефект се увеличава от това, че се нанасят икономически загуби. Теоретично ако тези животни не се бракуват и се използват за разплод хетерозиготни бици ще се загуби около 14% от всяко ново потомство в стадата, където се използват такива бици. Очевидно тези бици трябва да се елиминират. Разбира се възможно е да се получат генотипно и фенотипно здрави телета от хетерозиготни родители, но ако това се допусне рискът мутацията да се разпространи в популациите е много голям. На практика с цел изкореняване на този дефект са елиминирани от разплод всички потомци на два бика – на Whitehall Sultan от породата Шортхорн и на Prince Domino от Херефорд. За да се ускори изкореняването на дефекта при засегнатите породи се налага широко да се използва ДНК генотипизирането на всички одобрени за разплод животни особено на биците.
Литература.
1. DARWIN, CHARLES. 1882. A Naturalists Voyage Round the World. Pub.: John Murray, Albermark Et., London.
2. GREGORY, P. W., et al. 1953. Inheritance of Bovine Dwarfism and the Detection of Heterozygotes. Hilgardia, 22:13, p. 407-450.
3. E W. Stringam. Dwarfism in Beef Cattle. Canadian Journal of Comparative Medicine, 1958, 22, 11, 400-403.
4. T. A. OLSON. The Inheritance of Genetic Defects - Part 3, In: Cattle Genetics, 2008, http://www.braunviehcenter.com/cattle_genetics_part3.html
Дефицит на ензима кисела алфа-глюкозида (Pompe’s disease, Glycogen storage disease type II, or acid maltase deficiency).
Характеристика. Това заболяване принадлежи към групата на т. нар. “складови болести” (на англ. Glycogen storage diseases) и се наследява като автозомно-рецесивен признак. Когато рецесивният алел е в хомозиготно състояние в мускулните и нервните клетки се натрупва гликоген поради това, че ензимът лизозомна кисела алфа-глюкозида детерминиран от тези алели е с много ниска активност и поради това не може да го разгради до глюкоза, която е основен източник на енергия при много организми. Това заболяване е описано за първи път през 1932 г. при човека. Отложеният в клетките гликоген уврежда различни тъкани в тялото и причинява прогресивна мускулна слабост (миопатия) в сърцето, скелетната мускулатура, черния дроб и нервната система. При говедата и някои говедоподобни това е летално наследствено заболяване, което е диагностицирано при Брамана (порода зебу) и Шортхорна. За Брамана е характерна мутация в зона Е7 в промоторната част на гена и се среща при около 12% от популацията на тези животни. Открита е още една мутация – Е13, но тя е много рядка и е характерна за потомството на само едно внесено от чужбина животно. Поради това за в бъдеще всички внесени от чужбина животни ще бъдат предварително тествани чрез ДНК тест преди да бъде разрешено използването им за разплод. Заболяването се среща и при някои други видове животни като конете например.
Благодарение на изследванията на Dr. Peter Healy и неговата съпруга Dr. Julie Dennis от Elizabeth Macarthur Agricultural Institute (EMAI) при NSW вече се разполага с тест за двете мутации - E7 и E13. Тези тестове са много точни и откриват 100% от носителите им. Ако евентуално се открият нови мутации ще трябва да се разработят нови ДНК тестове. Обикновено за този тест се използват кръвни проби с антикоагулант EDTA, но ако има случай на фримартинизъм за изследване трябва да се използват косми с техните фоликули. Може да се използва и семенна течност от местни разплодници или при внос от чужбина. ABBA (Australian Brahman Breeders Association Ltd.) е организирала това изследване да се извършва в Genetic Disease Laboratory при EMAI за всички свои членове. Цената е $36,50 за едно животно.
Признаци. Клиничните признаци варират много и зависят изцяло от това в кои тъкани е отложен най-много гликоген. Най-често това се случва в мускулите и болните телета страдат от прогресивна мускулна атрофия. Признаците са най-очевидни, когато болното животно е под стрес например при отбиване, лошо хранене или пренаселеност. През по-късните етапи на болестта телетата забележимо забавят растежа си и може да бъдат намерени легнали на една старна и изпитват сериозни трудности при опит за ставане дори след като са загубили значителна част от теглото си. Телетата обикновено умират на възраст между 6 и 12 месеца. В много случаи се намират мъртви в трапчета, канавки или рекички и всичко изглежда като нещастен случай. Други телета може да умрат от внезапна сърдечна атака тъй като отложения гликоген е увредил сърдечния мускул. Когато е засегната нервната система телетата може да останат слепи дори от много ранна възраст.
Литература.
1. http://ghr.nlm.nih.gov/condition=pompedisease
2. http://www.agsdus.org/html/typeiipompe.htm,
3. http://www.agsdus.org/html/typeiipompe.htm,
4. http://www.agsdus.org/html/typeiipompe.htm,
5. "FDA Approval News for Myozyme".
6. http://www.fda.gov/bbs/topics/NEWS/2006/NEW01365.html. Retrieved 2009-12-16.
7. Burden of Proof: As Costs Rise, New Medicines Face Pushback; Insurers Limit Coverage To FDA-Approved Uses; $300,000 Drug Denied By Geeta Anand, The Wall Street Journal, September 18, 2007.
8. Canadian Common Drug Review Recommendations on Public Funding for Myozyme
9. Genzyme received broad approval in the European Union
9. Wagner KR (2007). "Enzyme replacement for infantile Pompe disease: the first step toward a cure". Neurology 68 (2): 88–9. doi:10.1212/01.wnl.0000253226.13795.40. PMID 17210887.
10. Chien YH; Lee, NC; Thurberg, BL; Chiang, SC; Zhang, XK; Keutzer, J; Huang, AC; Wu, MH et al. (2009). "Pompe disease in infants: improving the prognosis by newborn screening and early treatment". Pediatrics 124 (6): e1116–25. doi:10.1542/peds.2008-3667. PMID 19948615.
11. Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT (2001). "Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial.". Genetics in Medicine 3 (2): 132–138. doi:10.1097/00125817-200103000-00008. PMID 11286229. 12. 12. 12.http://journals.lww.com/geneticsinmedicine/pages/articleviewer.aspx?year=2001&issue=03000&article=00008&type=abstract.
13. Mendelsohn NJ; Messinger, YH; Rosenberg, AS; Kishnani, PS (2009). "Elimination of antibodies to recombinant enzyme in Pompe disease". N Engl J Med 360 (2): 194–195. doi:10.1056/NEJMc0806809. PMID 19129538.
14. Van der Ploeg AT (2010). "A randomized study of alucosidase alfa in late-onset Pompe disease". N Engl J Med 362 (15): 1396–1046. doi:10.1056/NEJMoa0909859. PMID 20393176.
15. Ausems MG, Verbiest J, Hermans MP, et al. (September 1999). "Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling". Eur. J. Hum. Genet. 7 (6): 713–6. doi:10.1038/sj.ejhg.5200367. PMID 10482961.
16. ABBA, Australian Brahman Breeders Association Ltd. Popes’s disease – commonly asked questions.
Наследствен дефицит на цинк при говедата (Bovine hereditary zinc deficiency). Наследственият дефицит на цинк при говедото (Bovine hereditary zinc deficiency -летален дефект A 46) е автозомно-рецесивно наследяващо се наследствено забляване с широк спектър на клинична манифестация и има летален характер ако не се лекува с високи дози цинк съдържащи препарати. Освен при говедата заболяването е установено при човека (acrodermatitis enteropathica) и мишките (lethal milk syndrome). При човека е установено, че гена отговорен за това заболяване е SLC39A4. Yuzbasyan-Gurkan and Barlett (2006) откриват аналога на SLC39A4 при говедото, който се характеризира с един уникален свързващ участък. Тази мутация е причина за прескачане на един от екзоните при транскрипцията на гена. Това е причина за образуване на дефектни трансмембранни домени отговорни за формирането на пори, през които се транспортира цинка в клетките. За първи път това заболяване е описано през 1964 г. при породата Черно-шарено фризийско говедо. Извършеният фамилен анализ на болните говеда е показал, че те имат един общ предшественик – бик на име Egbert NRS 13110 роден през 1932. Заболяването обаче най-често се свърза с друг бик на име Adema 21 van de Woudhoeve и поради това заболяването още се нарича болест на Адема ( “Adema disease”). Заболяването още е известно с имената Наследствена паракератоза и Наследствена хипоплазия на тимуса. По-късно заболяването е открито при Шортхорна в САЩ и Ангуса в Австралия. При човека, говедото и мишките основния дефект е невъзможност за ефективна абсорбция на цинка от гастроинтестиналния тракт. Цинкът е важен микроелемент и изпълнява важни функции в организма. Ролята на цинка като компонент на ензимите е съществена за осъществяване на биохимичните реакции. Например карбонанхидразата ускорява достигането на равновесието
CO2 + H2O ↔ HCO3- + H+
Правата реакция протича в тъканите, докато обратната се извършва в белите дробове и води до отделянето на СО2 от кръвта. Без действието на този фермент не би било възможно дишането. Цинкът е и в състава на хормона инсулин, който регулира съдържанието на захар в кръвта и усилва действието на хормоните на хипофизата. Следователно заболяването при човека и говедото се причинява от увреждането на различни генетични системи, които трябва да се проучат за да се изяснят метаболитните етапи, които са необходими за абсорбцията и дистрибуцията на цинка. За да се изясни клиниката, биохимизма и хистологичните промени изследователите от Мичиганския Щатски Университет изследват родословието на едно говедо болно от дефицит на цинк. Тук ще бъдат описани всички изменения, които са наблюдавани при болните животни от раждането им до навършване на 1 година.
Клинична манифестация. Клиничните признаци на заболяването започват да се проявяват, когато концентрацията на цинка в кръвта пада под 5 ррм, което при различните телета се случва между 16-я и 31-я ден след раждане, а животните са с живо тегло между 30 и 40 килограма. Наблюдаваните клинични признаци ще бъдат описани в хронологичен ред. Първите наблюдавани признаци са кожни лезии около муцуната на животните. Разпределението им е билатерално и симетрично, засягайки кожата периферно около ноздрите и латералните комисури на устната кухина с последващи изменения на интрамандибуларното пространство. Лезиите започват като сухи люспести петна, които в рамките на 24 часа се разцепват и се просмуква ексудат. Лезиите се намират на различно разстояние от муцуната на животните. На по-късен етап се образуват лезии около очите, каудалната част на темето и външната част на ушната мида. Четири от петте болни телета са образували лезии в областта на хълбоците, перианалната област и вентралната част на корема. Две от телетата са оставени да боледуват по-дълго време и са образували много суха и люспеста кожа по дължината на юголарните ямки, входа на гръдния кош и холката. В тези части обаче никога не е имало серозен ексудат. Диарията започва да се появява обикновено 3 дни след като концентрацията на цинка падне под 0,5 ррм. Изпражненията са били тъмнозелени и воднисти със сладникав метален мирис. Телетата показват летаргия и депресия и нямат желание да стават и да реагират на външни дразнения. Рефлексът за бозаене отслабва, но това не се дължи на лезиите около устата. Болните телета трудно обхващат с езика си биберона на бутилката с мляко. Вместо да сучат те извършват с езика си движения за дъвчене и дърпане, но срещу биберона. В резултат на това времето за хранене значително се удължава. Не се откриват лезии в устната кухина. Едновременно със затрудненото бозаене се забелязва и повишена саливация. Затруднинията в бозаенето и саливацията изчезват двадесет и четири часа след започване на лечението с цинкови препарати. Космената покривка на болните телета е суха и се забелязва значително намалена пигментация по върховете на космите в по-тъмно оцветените зони на кожата. Това е особено добре очертано в орбиталните зони. Полиозисът (загубата на пигмент в космите) е аналогична на “очилатата болест” при дефицит на елемента мед. Останалата част от космената покривка е сплъстена като груба тъкан, а върховете на космите изглеждат накъдрени. Много често засегнатите телета боледуват от пневмонии и гастро-ентеро-колити.
Лечение. Докато телетата са бозаели и по-късно получавали млекозаместител най-удачно е било лечението чрез добавяне към храната на цинков ацетат под формата на воден разтвор (10 ml вода + 3,4 g цинков ацетат). Тази доза е била добре понасяна от телетата. След навършване на една година на животните не е давано повече млекозаместител и цинковия ацетат е даван в количество 47,6 g в желатинови капсули. Трябва да подчертая, че всичко това е правено при експериментални условия. В условията на животновъдната практика това едва ли би било правено от фермерите, защото е свързано с финансови разходи и доста грижи за болните животни. Поради това единственото ефикасно средство за борба с това наследствено заболяване е откриване и елиминиране от разплод на хетерозиготните индивиди. За тази цел има разработен ДНК тест (Yuzbasyan-Gurkan and Barlett, 2006).
Литература.
Yuzbasyan-Gurkan V. and Barlett E. Identification of a unique splice site variant in SLC39A4 in bovine hereditary zinc deficiency, lethal trait A46: An animal model of acrodermatitis enteropathica. Genomics, 2006, 88, 4, 521-526.
1. L. Huang, J. Gitschier, A novel gene involved in zinc transport is deficient in the lethal milk mouse, Nat. Genet. 17 (1997) 292–297.
2. Online Mendelian Inheritance in Man. Johns Hopkins University, Baltimore, MD. MIM 201100 (edited 9/26/2003). World Wide Web URL: http://www.ncbi.nlm.nih.gov/omim/.
3. Online Mendelian Inheritance in Animals. Frank Nicholas, University of Sydney, Australia. MIA 000593.WorldWideWeb URL: http://omia.angis. org.au/retrieve.shtml?pid=1160.
4. E.A. McPherson, I.S. Beattie, G.B. Young, An inherited defect in Friesian calves, Nord. Vet. Med. 16 (1964) 535–540.
5. E. Andersen, T. Flagstad, A. Basse, E. Brummerstedt, Evidence of lethal trait A 46 in Black Pied Danish cattle of Friesian descent, Nord. Vet. Med. 22 (1970) 473–485.
6. D.W. Vogt, C.G. Carlton, R.B. Miller, Hereditary parakeratosis in Shorthorn beef calves, Am. J. Vet. Res. 49 (1988) 120–121.
7. R.W. Cook, P.A. Gill, Heritable zinc deficiency in Angus cattle, Proc. Annu. Conf. Aust. Soc. Vet. Pathol. (1993) 90.
Анхидротична ектодермална дисплазия при говедото (Anhidrotic Ectodermal Dysplasia in Cattle)
Фенотипна характеристика. Наследствените и конгенитални дисплазии при говедото са описани в редица клинични съобщения. Във връзка с това са описани широк спектър от наследствени ектодермални нарушения, които варират от нарушения в различните образувания на кожата до липса на космена покривка в различни части на тялото. Една рядка форма на X-свързана ектодермална дисплазия (MIA000543 [21]) показва изключително сходство със същото заболяване при човека (X-linked ED1 phenotype). Тази конгенитална X-свързана хипотрихоза, характеризираща се с липса на зъби и косми е описана при различни породи говеда [2, 4,23,29]. Други автори описват форми на хипотрихоза с по-слабо изразени клинични признаци. Напоследък при проучването на родословията на две фамилии говеда от породата Черношарен германски холщайн се установява хипотрихоза с олигодонтия [6,8]. Болните бици от тези родословия са имали генерализирана хипотрихоза, почти пълна липса на зъби и пълна липса на екринни назолабиални жлези [6]. Подобен фенотипен вариант на хипотрихоза, характеризираща се с почти пълна загуба на зъби е описана и при Червеношарения германски холщайн [8]. Хистологичното изследване на кожата е показало много тънък дермис с редки, атрофични космени фоликули и намалена плътност на потните жлези. Установена е също пълна липса на екринни назолабиални, трахеални и бронхиални жлези [8]. При болните мъжки животни и в двете фамилии е открит един общ предшественик намерен по майчина линия и е установено, че заболяването се наследява рецесивно моногенно свързано с половата Х-хромозома. Синдрома на плешивост при женските телета от породата Холщайн също е свързан с описаната хипотрихоза. При мъжките телета този автозомно-рецесивно наследяващ се признак е с летален ефект. Засегнатите телета се раждат с нормален външен вид, но след 1-2 месеца състоянието им се влошава и започват да се появяват обезкосмени петна по тялото. В засегнатите участъци кожата става по-дебела и набръчкана, а върха на ушите може да се накъдри. Телетата показват профузна саливация, измършавяват и болните женски телета умират на възраст 6-8 месеца. Подобен синдром е описан при породата безрог Херефорд и е известен под различни имена като конгенитална анемия, дискератоза и прогресираща алопеция. Характерен е и за двата пола. Още при раждане се установява нискостепенна анемия, която постепенно прогресира с напредване на възрастта. Около муцуната и краищата на ушите се появява алопеция, ненормално къдрави косми и хиперкератоза, които се уголемяват със съзряването на животните. По-късно кожата по описаните места се набръчква и се появяват невни признаци. Телетата имат диария и умират преди да са навършили 6 месеца. Съвремените молекулярно генетични методи позволяват бързо откриване на това наследствено заболяване при говедата.
Мутационен анализ. За извършване на мутационен анализ на кодиращите части на ген ED1 са синтезирани двойки праймери за амплификация на изолираната ДНК от членовете на описаните фамилии чрез PCR [5]. След това всеки индивидуален ED1 екзон е изследван за мутации чрез секвениране и сравнение с контролна говежда ДНК. За потвърждаване на откритите геномни мутации са извършени и RT-PCR експерименти на ниво ED1 mRNA. Амплифицираните RT-PCR продукти след това са отново секвенирани. При една от фамилиите на Черношарения германски холщайн PCR анализа на 12 животни показва, че ED1 екзон 3 не може да бъде амплифициран при болните животни, което е доказателство за наличие на геномна делеция в този екзон. За разлика от първата фамилия във втората (от породата Червеношарен германски холщайн) е открита точкова мутация в ген ED1. Секвенирането на PCR продуктите, принадлежащи на едно от болните мъжки животни и неговата майка показва, че болното теле е хемизиготно по база G докато майката е хетерозиготна по бази T/G при втора позиция на интрон 8 [7]. И при двете фамилии всички други ED1 екзони са без изменения и не показват никакъв полиморфизъм между болните и здрави животни [5, 7]. Изследването чрез RT-PCR е потвърдило, че ED1 mRNA от всички болни животни от фамилиите на Черношарения германски холщайн имат увреден екзон 3 и че три женски животни са хетерозитотни носители на патологична Х-свързана наследствена мутация [5, 6]. RT-PCR и cDNA секвенирането демонстрира, че една точкова мутация в 50-я свързващ участък на екзон 8b може да засегне правилното свързване на двата гена ED1-A1 и ED1-A2. При едно от болните животни от породата Червеношарен германски холщайн използването на защитните вътрешни свързващи донорни и акцепторни части в рамкита на екзон 8 води до получаването на единична траскрипционна загуба на 51 или 45 bp, което има отношение към фукцията на транскрипцията на гени ED1-A1 или ED1-A2 съответно [7]. При един болен човек (human ED1 patient) една мутация в 50-я свързващ участък на екзон 8b (IVS8C5G > A) на ген ED1 може би е причина за увреждането само на транскрипта ED1-A1 [25]. Тъй като анализите посочват наличието на един важен свързващ енхенсер в началото на интрон 8 според авторите може би си струва да се изследват експериментално последователностите на транскрипциония процес при този човек. Делецията в екзон 3 на ген ED1 е причина за загуба на определен брой нуклеотиди, което води до скъсяване на протеина, който губи колагеноподобен тримеризационен (полимер образуван от три молекули на мономера) домен, а също и функционално важният TNF-подобен сигнален домен на ектодисплазин A1 и A2 протеините. На протеиново ниво се предполага, че мутацията в свързващия участък е резултат от вътрешна делеция в голям участък на функционално важния TNF-подобен сигнален домен на ектодисплазин 1. В резултат на описаните генетични находки X-свързания наследствен фенотип при болните говеда с хипотрихоза, олигодонтия, Х-свързания дефект ED1, който при говедата се означава като S143 и липсата на екринни жлези се счита, че се причинява от мутации в ген ED1 и поради това се нарича Анхидротична ектодермална дисплазия (ED1) при говедата.
Литература.
Основен източник: Cord DRÖGEMÜLLER, Ottmar DISTL, Tosso LEEB. X-linked anhidrotic ectodermal dysplasia (ED1) in men, mice, and cattle. Genet. Sel. Evol. 35 (Suppl. 1) (2003) S137–145.
[1] Bayés M., Hartung A.J., Ezer S., Pispa J., Thesleff I., Srivastava A.K., Kere J. The anhidrotic ectodermal dysplasia gene (EDA) undergoes alternative splicing and encodes ectodysplasin-A with deletion mutations in collagenous repeats, Hum. Mol. Genet. 7 (1998) 1661–1669.
[2] Braun U., Ansari H.A., Hediger R., Süss U., Ehrensperger F. Hypotrichosis and oligodontia associated with a chromosomal Xq-deletion in a Simmental/Red Holstein crossbreed [German], Tierärztl. Prax. 16 (1988) 39–44.
[3] Chen Y., Molloy S.S., Thomas L., Gambee J., Bachinger H.P., Ferguson B. Zonana J., Thomas G.,Morris N.P., Mutations within a furin consensus sequence block proteolytic release of ectodysplasin-A and cause X-linked hypohidrotic ectodermal dysplasia, Proc. Natl. Acad. Sci. USA 98 (2001) 7218–7223.
[4] Drieux H., Priouzeau M., Thiéry G., Priouzeau M.L. Hypotrichose congénitale avec anodontie, acérie et macroglossie chez le veau, Rec. Med. Vet. 126 (1950) 385–399.
[5] Drögemüller C., Distl O., Leeb T., Partial deletion of the bovine ED1 gene causes anhidrotic ectodermal dysplasia in cattle, Genome Res. 11 (2001) 1699–1705.
[6] Drögemüller C., Kuiper H., Peters M., Guionaud S., Distl O., Leeb T., Congenital hypotrichosis with anodontia in cattle: A genetic, clinical and histological analysis, Vet. Dermatol. 13 (2002) 307–313.
[7] Drögemüller C., Peters M., Pohlenz J., Distl O., Leeb T., A single point mutation within the ED1 gene disrupts correct splicing at two different splice sites and leads to anhidrotic ectodermal dysplasia in cattle, J. Mol. Med. 80 (2002) 319–323.
[8] Drögemüller C., Kuiper H., Leeb T., Peters M., Pohlenz J., Distl O., Congenital hypotrichosis and oligodontia in cattle [German], Tierärztl. Prax. 31 (2003), 70–76.
[9] Elomaa O., Pulkkinen K., Hannelius U.,Mikkola M., Saarialho-Kere U., Kere J., Ectodysplasin is released by proteolytic shedding and binds to the EDAR protein, Hum. Mol. Genet. 10 (2001) 953–962.
[10] Ezer S., Bayés M., Elomaa O., Schlessinger D., Kere J., Ectodysplasin is a collagenous trimeric type II membrane protein with a tumor necrosis factor-like domain and co-localizes with cytoskeletal structures at lateral and apical surfaces of cells, Hum. Mol. Genet. 8 (1999) 2079–2086.
[11] Ferguson B.M., Brockdorff N., Formstone E., Ngyuen T., Kronmiller J.E., Zonana J., Cloning of Tabby, the murine homolog of the human EDA gene: evidence for a membrane-associated protein with a short collagenous domain, Hum. Mol. Genet. 6 (1997) 1589–1594.
[12] Headon D.J., Overbeek P.A., Involvement of a novel Tnf receptor homologue in hair follicle induction, Nat. Genet. 22 (1999) 370–374.
[13] Headon D.J., Emmal S.A., Ferguson B.M., Tucker A.S., Justice M.J., Sharpe P.T., Zonana J., Overbeek P.A., Gene defect in ectodermal dysplasia implicates a death domain adapter in development, Nature 414 (2001) 913–916.
[14] Kere J., Srivastava A.K., Montonen O., Zonana J., Thomas N., Ferguson B., Munoz F., Morgan D., Clarke A., Baybayan P., Chen E.Y., Ezer S., Saarialho- Kere U., de la Chapelle A., Schlessinger D., X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein, Nat. Genet. 13 (1996) 409–416.
[15] Koppinen P., Pispa J., Laurikkala J., Thesleff I., Mikkola M.L., Signalling and subcellular localization of the TNF receptor Edar, Exp. Cell. Res. 269 (2001) 180–192.
[16] Kumar A., EbyM.T., Sinha S., Jasmin A., Chaudhary P.M., The ectodermal dysplasia receptor activates the nuclear factor-kappaB, JNK and cell death pathways and binds to ectodysplasin, J. Biol. Chem. 276 (2001) 2668–2677.
[17] Locksley R.M., Killeen N., Lenardo M.J., The TNF and TNF receptor superfamilies: integrating mammalian biology, Cell 104 (2001) 487–501.
[18] Mikkola M.L., Pispa J., Pekkanen M., Paulin L., Nieminen P., Kere J., Thesleff I., Ectodysplasin, a protein required for epithelial morphogenesis, is a novel TNF homologue and promotes cell-matrix adhesion, Mech. Dev. 88 (1999) 133–146.
[19] Monreal A.W., Zonana J., Ferguson B., Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations, Am. J. Hum. Genet. 63 (1998) 380–389.
[20] Monreal A.W., Ferguson B.M., Headon D.J., Street S.L., Overbeek P.A., Zonana J.,Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia, Nat. Genet. 22 (1999) 366–369.
[21] Nicholas F.W., Genetic databases: online catalogues of inherited disorders, Rev. Sci. Tech. 17 (1998) 346–350.
[22] Online Mendelian Inheritance inMan,OMIM(TM).McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD) (2000) http://www.ncbi.nlm.nih.gov/omim/
[23] Rieck G.W., Hypotrichie-Hypodontie-Syndrom beim Rind [German], Dtsch. Tierärztl. Wschr. 92 (1985) 328–329.
[24] Schmidt-Ullrich R., Aebischer T., Hulsken J., Birchmeier W., Klemm U., Scheidereit C., Requirement of NF-kappaB/Rel for the development of hair follicles and other epidermal appendices, Development 128 (2001) 3843–3853.
[25] Schneider P., Street S.L., Gaide O., Hertig S., Tardivel A., Tschopp J., Runkel L., Alevizopoulos K., Ferguson B.M., Zonana J., Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A, J. Biol. Chem. 276 (2001) 18819–18827.
[26] Selmanowitz V.J., Ectodermal dysplasias in cattle – Analogues in man, Br. J. Dermatol. 84 (1970) 258–265.
[27] Srivastava A.K., Pispa J., Hartung A.J., Du Y., Ezer S., Jenks T., Shimada T., Pekkanen M., Mikkola M.L., Ko M.S.H., Thesleff I., Kere J., Schlessinger D., The tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-A) with collagenous domains, Proc. Natl. Acad. Sci. USA 94 (1997) 13069–13074.
[28] SrivastavaA.K., DurmowiczM.C., Hartung A.J., Hudson J., Ouzts L.V., Donovan D.M., Cui C.Y., Schlessinger D., Ectodysplasin-A1 is sufficient to rescue both hair growth and sweat glands in tabby mice, Hum. Mol. Genet. 10 (2001) 2973–2981.
[29] Wijeratne W.V.S., O’Toole D., Wood L., Harkness J.W., A genetic, pathological and virological study of congenital hypotrichosis and incisor anodontia in cattle, Vet. Rec. 122 (1988) 149–152.
[30] Yan M., Wang L.C., Hymowitz S.G., Schilbach S., Lee J., Goddard A., de Vos A.M., Gao W.Q., Dixit V.M., Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors, Science 290 (2000) 523–527.
[31] Yan M., Zhang Z., Brady J.F., Schilbach S., Fairbrother W.J., Dixit V.M., Identification of a novel death domain-containing adaptor molecule for ectodysplasin-A receptor that is mutated in crinkled mice, Curr. Biol. 12 (2002) 409–413.
Конгенитална синдактилия при говедото (Congenital syndactyly in cattle). Много наследствени малформации при домашните животни са аналожни с подобни на тях при човека и поради това са много подходящи за модели, чрез които да се изучава патогенезата на тези заболявания при различни фенотипове с идентична молекулна основа. Конгениталната синдактилия при говедото засяга само пръстите на крайниците, което може да се изразява чрез сливане или неразделяне на двата пръста на крайниците. Този феномен често наподобява дисталната част на крайниците при еднокопитните и поради това това заболяване се нарича още “крак на муле” (mulefoot). Синдактилията при говедото фенотипно се експресира различно като най-често засягат предните крайници (един или два), но когато са засегнати и четирите крайника тежеста на дефекта е в посока десен-ляв като по-тежко са увредени предните крайници. Това заболяване се изразява основно в сливане на хоризонтално сливане на проксималните фаланги на пръстите. Някои автори дори приемат това хоризонтално сливане като основен критерий за диференциране на наследтвената форма от придобитата. Счита се, че синдактилията при говедото се наследява като автозомно-рецесивен признак с непълна пенетрантност при много породи говеда в различни държави по света. С най-висока честота е установена при породите Холщайн и Ангус. Чрез генетично кантиране е локализиран локуса отговорен за този дефект върху 15 хромозома на говедото. Този регион от говеждата хромозома е хомоложен със определен сегмент от хромозома 2 при мишките, в който се намира гена, кодиращ рецептор за липопротеин с ниска плътност (Lrp4), допълнително определен като ген 7, кодиращ множествен домейн на епидермалния растежен фактор (Megf7). Хомозиготните Lrp4-дефицитни мишки са с изостанало развитие и пълна пенетрантност на полисиндактилията на предните и задни крайници. При човека синдактилията се представя най-често с конгенитална малформация на ръцете, която се характеризира с различно по степен сливане на меките тъкани на пръстите с или без сливане на костите. Засега са открити два гена, които да кодират синдактилията при човека - HOXD13 и GJA1. Различната фенотипна експресия на заболяването предполага наличието на повече гени.
До 2006 година бяха известни само 2 алела на ген LRP4 отговорни за синдактилията при породите Холщайн и Ангус (9, 10). През 2007 година бяха открити още 4 мутации на този ген, което обяснява различната фенотипна проява на заболяването (Drögemüller и др.). Мутациите се представят като замествания на едни нуклеотидни бази с други. Например при 36 телета от породата Холщайн са открити две последователни замествания на нуклеотиди в екзон 33 на ген LRP4 (c.4863_4864delCGinsAT), което от своя страна е довело до замяна на една аминокиселина с друга (p. [Asn1621Lys; Gly1622Cys]). При две телета от породата Ангус са открити в хомозиготно състояние две единични нуклеотидни замени в първата база на интрон 37 (c.5385+1G>A).
Литература
1. Patterson DF, Haskins ME, Jezyk PF, Giger U, Meyers-Wallen VN, Aguirre G, Fyfe JC, Wolfe JH: Research on genetic diseases: reciprocal benefits to animals and man. J Am Vet Med Assoc 1988, 193:1131-1144.
2. Leipold HW, Adrian RW, Huston K, Trotter DM, Dennis SM, Guffy MM: Anatomy of hereditary bovine syndactylism. I. Osteology. J Dairy Sci 1969, 52:1422-1431.
3. Drögemüller C, Distl O: Genetic analysis of syndactyly in German Holstein cattle.
Vet J 2006, 171:120-125.
4. Charlier C, Farnir F, Berzi P, Vanmanshoven P, Brouwers B, Vromans H, Georges M: Identity-by-descent mapping of recessive traits in livestock: application to map the bovine syndactyly locus to chromosome 15. Genome Res 1996, 6:580-589.
5. Johnson EB, Hammer RE, Herz J: Abnormal development of the apical ectodermal ridge and polysyndactyly in Megf7-deficient mice. Hum Mol Genet 2005, 14:3523-3538.
5. Simon-Chazottes D, Tutois S, Kuehn M, Evans M, Bourgade F, Cook S, Davisson MT, Guenet JL: Mutations in the gene encoding the low density lipoprotein receptor LRP4 cause abnormal limb development in the mouse. Genomics 2006, 87:673-677.
6. Herz J: The LDL receptor gene family: (un)expected signal transducers in the brain.
Neuron 2001, 29:571-581.
7. Schwabe GC, Mundlos S: Genetics of congenital hand anomalies. Handchir Mikrochir Plast Chir 2004, 36:85-97.
8. Duchesne A, Gautier M, Chadi S, Grohs C, Floriot S, Gallard Y, Caste G, Ducos A, Eggen A: Identification of a doublet missense substitution in the bovine LRP4 gene as a candidate causal mutation for syndactyly in Holstein cattle. Genomics 2006, 88:610-621.
9. Johnson EB, Steffen DJ, Lynch KW, Herz J: Defective splicing of Megf7/Lrp4, a regulator of distal limb development, in autosomal recessive mulefoot disease. Genomics 2006, 88:600-609.
10. Leipold HW, Schmidt GL, Steffen DJ, Vestweber JG, Huston K: Hereditary syndactyly in Angus cattle. J Vet Diagn Invest 1998, 10:247-254.
11. Wöhlke A, Kuiper H, Distl O, Drögemüller C: The bovine aristaless-like homeobox 4 (ALX4) as a candidate gene for syndactyly. Cytogenet Genome Res 2006, 115:123-128.
12. Goldstein JL, Hobbs HH, Brown MS: Familial hypercholesterolemia. In Metabolic and Molecular Bases of Inherited Disease. Edited by Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B. McGraw-Hill, New York; 2001:2863-2913.
13. Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, Eddy SR, Sonnhammer EL, Bateman A: Pfam: clans, web tools and services. Nucleic Acids Res 2006, 34:D247-251.
14. Pfam [http://www.sanger.ac.uk/Software/Pfam/] webcite
15. Ramensky V, Bork P, Sunyaev S: Human non-synonymous SNPs: server and survey.
Nucleic Acids Res 2002, 30:3894-3900.
16. PolyPhen http://genetics.bwh.harvard.edu/pph/ webcite
17. Ng Pc, Henikoff S: Predicting deleterious amino acid substitutions. Genome Res 2001, 11:863-874.
18. SIFT [http://blocks.fhcrc.org/sift/SIFT.html] webcite
19. Hattori H, Nagano M, Iwata F, Homma Y, Egashira T, Okada T: Identification of recurrent and novel mutations in the LDL receptor gene in Japanese familial hypercholesterolemia. Hum Mutat 1999, 14:87.
20. Esser V, Russell DW: Transport-deficient mutations in the low density lipoprotein receptor. Alterations in the cysteine-rich and cysteine-poor regions of the protein block intracellular transport. J Biol Chem 1988, 263:13276-13281.
21. Nakayama M, Nakajima D, Nagase T, Nomura N, Seki N, Ohara O: Identification of high-molecular-weight proteins with multiple EGF-like motifs by motif-trap screening.
Genomics 1998, 51:27-34.
22. Heath KE, Gahan M, Whittall RA, Humphries SE: Low density lipoprotein receptor gene (LDLR) world-wide website in familial hypercholesterolaemia: update, new features and mutation analysis. Atherosclerosis 2001, 154:243-246.
ВЪПРОСНИК ЗА НАСЛЕДСТВЕНИТЕ БОЛЕСТИ ПРИ ГОВЕДОТО
(Верните текстове да се подчертаят!)
Наследствен дефицит в активността на ензима Алфа - манозидаза (Alpha-Mannosidosis).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно.
За кои породи е характерно - Ангус, Мъри грей, Галоуей, Херефорд, Джерсей, БКГ.
Какъв ДНК тест се използва за откриване на хетерозигон(тните индивиди – обикновен PCR, директен PCR, RFLP.
Какви се клиничните признаци - умират скоро след раждане, а тези които все пак оцелеят показват тежко, прогресиращо неврологично заболяване, което се характеризира с тремор на главата, атаксия и агресия, диария, пневмония.
Наследствен дефицит в активността на ензима Бета - манозидaза (Beta-Mannosidоsis).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно.
За кои породи е характерно - Ангус, Мъри грей, Галоуей, Херефорд, Джерсей, БКГ, Салерс.
Какви са клиничните признаци - болните телета не растат нормално и показват клатещи движения на главата и тремор на други телесни части. Лицевите деформации включват прогнатизъм (издаване на горната или долната челюст), тесни цепки на клепачите и насочени назад уши, бъчвообразна постановка на крайниците.
Артрогрипозис мултиплекс (Arthrogryposis Multiplex - АМ).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно.
За кои породи е характерно - Ангус, Мъри грей, Галоуей, Херефорд, Джерсей, БКГ, Салерс.
Какви са клиничните признаци - телетата се раждат или мъртви или са умирали скоро след раждане. Гърбът и краката са извити или изкривени в ставите и често са фиксирани в тази позиция. Предните крайници са свити и повдигнати или изпънати. Телетата са малки по размер поради слабо развитата мускулатура. Понакога е има разцепване на носната част или на горното небце. Метеоризъм на червата и подуване на предстомашията.
хезионен дефицит на говеждите лимфоцити (Bovine Lymphocyte Adhesion Deficiency - BLAD).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно.
За кои породи е характерно – Холщайн, Ангус, Мъри грей, Галоуей, Херефорд, Джерсей, БКГ, Салерс.
Какви са клиничните признаци - клиничните признаци на това заболяване се проявяват на възраст 1-2 седмици и се характеризират с язви по устната лигавица, тежък периодонтит, липса на зъби, хронична пневмония и повтаряща се хронична диария. Хематологичните изследвания показват неутрофилия (често >100,000) без изместване наляво, лимфоцитоза и моноцитоза. Биохимичните изследвания се характеризират с хипоалбуминемия, хиперглобулинемия, ниски стойности на креатинина, уреята и глюкозата. Болните телета умират на възраст 2-4 месеца поради инфекциозни усложнения. Страдат от кифоза, артрогрипозис мултиплекс, куцота.
Каква е честотата на разпространение на заболяването – 5%, 10%, 24%.
Каква е молекулната същност на BLAD - точкова мутация (замяна на аденин с гуанин) в позиция 383 на ген CD18, ген CD11, ген CD12.
Кой бик е основният разпространител на заболяването - Osborndale Ivanhoe, други бици.
Наследствен дефицит на ензима уридин монофосфат синтетаза (Deficiency of Uridine Monophosphate Synthetase – DUMPS).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно.
За кои породи е характерно – Холщайн, Ангус, Мъри грей, Галоуей, Херефорд, Джерсей, БКГ, Салерс.
Какви е съдбата на хомозиготните телета - хомозиготните по мутантния алел ембриони не оцеляват до раждане и обикновено умират през ранен етап на бременоста. Ембрионите вероятно се абортират или реабсорбират около 40-я ден от бремеността и това предизвиква повтарящи се маточни кръвоизливи. Оцеляват, достигат репродуктивна възраст и имат висока млечност.
Каква е молекулната същност на DUMPS - в клетките на бозайниците уридин монофосфат синтетазата (UMPS) е ензим отговорен за конверсията на оротовата киселина до уридин монофосфат, който от своя страна е важен компонент на пиримидин нуклеотидите. Този ензим всъщност има две ензимни функции: оротова фосфорибозил трансфераза и оротидин монофосфат декарбоксилаза, която кореспондира с последните два етапа на пиримидин синтетазата.
Каква мутация е причина за това заболяване - мутацията С-Т (т.е. заместване на цитозин с тимин) в гена кодиращ уридин монофосфат синтетазата при кодон 405 от екзон 5 на хромозома 1 води до образуване на стоп кодон и вследствие на това се образува функционално увреден ензим. Точкова мутация (замяна на аденин с гуанин).
В кои държави е открито заболяването - наличие на мутация в гена кодиращ уридин монофосфат синтетазата е открита още в Аржентина, Унгария, Тайван и Турция, Полша и Индия.
Комплескна вертебрална малформация (Сomplex vertebral malformation - CVM).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно.
За кои породи е характерно – Холщайн, Ангус, Мъри грей, Галоуей, Херефорд, Джерсей, БКГ, Салерс.
Какви са морфологичните изменения при болните телета - деформирани прешлени в шийната, торакалната и лумбалната област на на гръбначния стълб, аномалии в стернума и ребрата, ниско живо тегло и латерална ротация на ставите в метакарпалната и метатарзалната област. При изследване на сърцето се установява дясностранна хипертрофия, дефект в преградата между предсърдията и много фетуси с CVM се абортират на 159-я ден от бремеността, а други се раждат преждевременно или мъртви. Оцеляват и се развиват нормално.
Каква мутация е причина за това заболяване - молекулната основа на CVM е заместване на гуанин с тимин (G-T) в ген 3 (SLC35A3), кодон 559 на генна фамилия 35, хромозома 3, който кодира UDP-N-ацетилглюкозамин транспортера. Хромозомна аберация.
В кои държави е открито заболяването - Най-напред CVM е открита при телета от породата Датски Холщайн през 2000 година и след това наличието на CVM носители са докладвани и в други държави като Швеция, Дания, Чехия и Полша, България.
Наследствен дефицит на фактор XI (Factor XI – plasma thromboplastin antecedent deficiency in cattle).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно.
За кои породи е характерно – Холщайн, Ангус, Мъри грей, Галоуей, Херефорд, Джерсей, БКГ, Салерс.
Каква е клиничната манифектация на заболяването - удължено време за коагулация на кръвта, сепсис.
Каква мутация е причина за това заболяване - инсерция с размер 77 чифта бази (AT (A)29 TAAAG (A)27 GAATTATTAATTCT) в рамките на екзон 12 на ген FXI. Робертсонова транслокация 1/29.
Наследствена тромбопатия при породата Симентал (Platelet Bleeding Disorder, Simental hereditary thrombopathy).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно.
За кои породи е характерно –Галоуей, Херефорд, Джерсей, БКГ, Салерс, Симентал.
Каква е клиничната манифектация на заболяването - удължено време за коагулация на кръвта, сепсис, заболяването се характеризира с спонтанен епистаксис (кръвотечение от носа), повърхностни хематоми, продължителни кръвотечения при нараняване или дори най-леки манипулации като татуиране или инжекции. Наблюдавани са също спонтанни мукозни кръвоизливи и хематурия. Наблюдавани са тежки кръвоизливи от носа особено при студено време.
Каква мутация е причина за това заболяване - инсерция с размер 77 чифта бази (AT (A)29 TAAAG (A)27 GAATTATTAATTCT) в рамките на екзон 12 на ген FXI. Робертсонова транслокация 1/29. Нуклеотидна замяна Т-С в позиция 701, което от своя страна води до заместване на аминокиселината пролин с левцин, а това е причина за структорна промяна във втори регион (SCR2) на каталитичния домейн на специфичния протеин отговорен за агрегацията на тромбоцитите.
Протопорфирия (Protoporphyria, Congenital Erythropoietic Porphyria, Pink tooth, Osteohemochromatosis).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно.
За кои породи е характерно – млечен тип говеда, месодайни говеда.
Каква е клиничната манифектация на заболяването - Наблюдавани са тежки кръвоизливи от носа особено при студено време, червеникави на цвят зъби, кости и урина, които перзистират през целия живот на болните животни. Наследственият ензимен дефект е причина за дефицит в активността на ензима уропорфириноген III косинтетаза, която е важна част от порфирин-хем биосинтезата. Урината на засегнатите животни съдържа високи количества на копротопорфирин I и уропорфирин I и поради това цвета й е кехлибарен или червено-кафяв. Костите, урината и зъбите (особено млечните зъби) флуоресцират розово, когато се облъчат с ултравиолетова светлина отблизо. Продължителното излагане на слънчева светлина, поради фотосензибилизацията, причинява типични лезии с хиперемия, образуване на везикули и повърхностни некрози на непигментираните участъци на кожата.
Каква мутация е причина за това заболяване - Робертсонова транслокация 1/29. Наследственият ензимен дефект е причина за дефицит в активността на ензима уропорфириноген III косинтетаза, която е важна част от порфирин-хем биосинтезата. Урината на засегнатите животни съдържа високи количества на копротопорфирин I и уропорфирин I и поради това цвета й е кехлибарен или червено-кафяв.
Какво е разпространението на заболяването – ограничено, в целия свят.
Прогресивна дегенеративна миелоенцефалопатия при говедото (Bovine progressive degenerative myeloencephalopathy - BPDME, weaver syndrome).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно, фамилно разпространение.
За кои породи е характерно – млечен тип говеда, месодайни говеда.
Каква е клиничната манифектация на заболяването - 1) едновременна билатерална атаксия на задните крайници и проявена дисметрия (неспособност или нарушена способност за точен контрол на степента на движенията при мускулни действия. Също невъзможност за точно определяне на разстояния), когато животните са на възраст 5-8 месеца; 2) неадекватни проприоцептивни отговори (това са рецептори, които се намират главно в мускулите, сухожилията, ставите и вътрешното ухо, които определят движенията или позицията на тялото и крайниците при отговор на дразнене възникнало вътре в организма), атаксия при четирите крайника и прогресивна парапареза; 3) нормални спинални рефлекси и запазена функция на краниалните нерви и липса на тежка мускулна атрофия; 4) фамилна връзка на разпространение при установяване на посочените признаци.
За кои породи е характерно –Галоуей, Херефорд, Джерсей, Кафявото Швейцарско говедо (още се нарича Сивокафяво алпийско говедо).
На каква възраст се проявява заболяването - се проявяват обикновено на възраст между 5 и 8 месеца. През следващите 12-18 месеца болните животни проявяват прогресираща атаксия с прогресираща деориентация на локомоторната функция, която засяга най-напред задните крайници. На възраст 18-24 месеца повечето болни животни развиват тежка атаксия на задните крайници с явно отслабени проприоцептивни рефлекси. През по-късните стадии на заболяването проприоцептивния дефицит става още по-очевиден дори когато животните са прави. Слепите животни болни от BPDME показват по-силно изразена атаксия, дисметрия, проприоцептивен дефицит и люлеещи се задни крайници, когато са в покой. При тежките случаи се забелязва умерена и генерализирана загуба на телесна маса с умерена атрофия на бедрената мускулатура.
Джуджевидност при говедата за месо (Dwarfism).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно, фамилно разпространение.
За кои породи е характерно – млечен тип говеда, месодайни говеда.
Форми на заболяването:
- Изключителни ("Snorters")
- Компактна (компресирана) форма
- Тип дребосък, дългоглави телета (Miscellany "Longheads", "Pin-heads", Midgets)
Нанася ли заболяването икономически загуби – да, не.
Дефицит на ензима кисела алфа-глюкозида (Pompe’s disease, Glycogen storage disease type II, or acid maltase deficiency).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно, фамилно разпространение.
За кои породи е характерно – млечен тип говеда, месодайни говеда.
Какъв патофизиологичен тип е това заболяване – тип “складови болести”, възпалителни, дегенеративни.
Признаци - телетата забележимо забавят растежа си и може да бъдат намерени легнали на една старна и изпитват сериозни трудности при опит за ставане дори след като са загубили значителна част от теглото си, преплетена походка, атаксия.
Наследствен дефицит на цинк при говедата (Bovine hereditary zinc deficiency).
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно, фамилно разпространение.
За кои породи е характерно – млечен тип говеда, месодайни говеда.
Признаци:
- кожни лезии около муцуната на животните
- лезиите започват като сухи люспести петна, които в рамките на 24 часа се разцепват и се просмуква ексудат. Лезиите се намират на различно разстояние от муцуната на животните.
- на по-късен етап се образуват лезии около очите, каудалната част на темето и външната част на ушната мида.
- лезии в областта на хълбоците, перианалната област и вентралната част на корема.
- диарията започва да се появява обикновено 3 дни след като концентрацията на цинка падне под 0,5 ррм. Изпражненията са тъмнозелени и воднисти със сладникав метален мирис. Телетата показват летаргия и депресия и нямат желание да стават и да реагират на външни дразнения.
- рефлексът за бозаене отслабва, но това не се дължи на лезиите около устата. Болните телета трудно обхващат с езика си биберона на бутилката с мляко.
Има ли лечение това заболяване – да, не.
Анхидротична ектодермална дисплазия при говедото (Anhidrotic Ectodermal Dysplasia in Cattle)
Как се наследява заболяването - автозомно-рецесивно, доминантно, кодоминантно, интермедиерно, фамилно разпространение, свързано с пола.
За кои породи е характерно – Черношарен германски холщайн, Червеношарения германски холщайн, Червено датско говедо, Англер.
В кой ген се намира мутацията, причиняваща това заболяване - ED1, ED2, ED3, ED4
Признаци:
- хипотрихоза
- олигодонтия
- кетоза
- ацидоза
Конгенитална синдактилия при говедото (Congenital syndactyly in cattle).
Как се наследява заболяването - автозомно-рецесивно, автозомно-рецесивен признак с непълна пенетрантност, доминантно, кодоминантно, интермедиерно, фамилно разпространение, свързано с пола.
За кои породи е характерно – Холщайн, Червено датско говедо, Англер, Ангус.
В кой ген се намира мутацията, причиняваща това заболяване - ген LRP4, ген LRP5, ген LRP6, ген LRP7.
Признаци:
- кетоза
- ацидоза
- сливане или неразделяне на двата пръста на крайниците
- засегнати са и четирите крайника като тежеста на дефекта е в посока десен-ляв като по-тежко са увредени предните крайници

